La chiralità è la proprietà geometrica di un insieme di punti o atomi nello spazio, o di oggetti solidi, di non essere sovrapponibili alla loro immagine speculare.[1] Questi strutture, definite chirali, hanno la caratteristica peculiare di essere prive di elementi di simmetria del secondo ordine, ossia un piano di simmetria, un centro di inversione, o un asse di roto-riflessione.[2]
La definizione di chiralità, dal greco cheir che significa “mano”, si deve a Lord Kelvin che la enunciò nel corso delle “Baltimore Lectures”, una serie di lezioni tenute alla Johns Hopkins University di Baltimora a partire dal primo ottobre del 1884, e pubblicate nel 1904.[3]
L’ambiente che ci circonda è ricco di oggetti chirali: le nostre mani ne sono l’esempio per eccellenza, ma ce ne sono moltissimi altri, dal guscio di una chiocciola sino a una galassia a spirale. In chimica, e in special modo in chimica organica, la chiralità è una proprietà di importanza primaria, in quanto molecole come i carboidrati, molti amminoacidi, nonché moltissimi farmaci, sono chirali.[4]
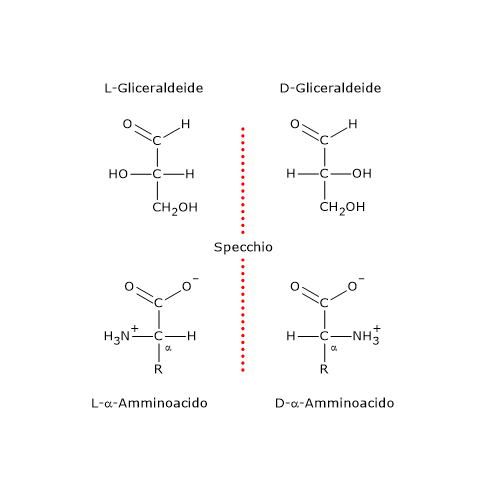
Le molecole chirali possono esistere in due forme, l’una immagine speculare dell’altra e non sovrapponibili, ossia, non esiste una combinazione di rotazioni o traslazioni sul piano del foglio che consenta la loro sovrapposizione. Queste molecole sono dette enantiomeri, dal greco enántios che significa “contrario” e meros che significa “parte”.[5]
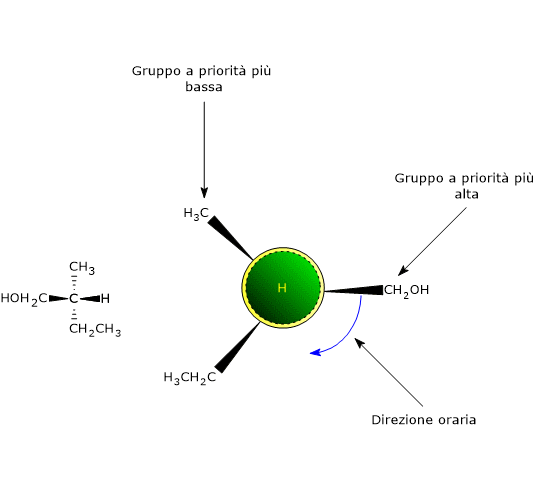
La causa più comune di chiralità in una molecola è la presenza di un centro chirale centro di chiralità, anche detto centro asimmetrico, cioè un atomo che leghi un insieme di ligandi disposti nello spazio in modo che la molecola risultante possa esistere come due enantiomeri.
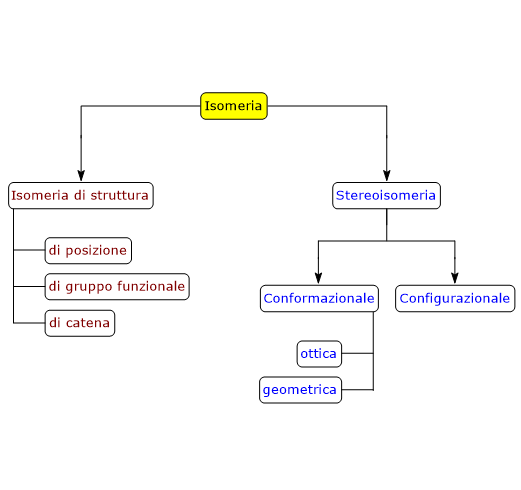
Gli enantiomeri a loro volta sono un tipo di stereoisomeri, i quali possono essere definiti come isomeri che hanno lo stesso numero e tipo di atomi e legami, ma che differiscono nell’orientamento spaziale degli atomi.[2]
Indice
Enantiomeri
Due enantiomeri di una molecola chirale, non essendo sovrapponibili, sono composti differenti. In che cosa differiscono?
Ciascuna coppia di enantiomeri possiede identiche proprietà fisiche e chimiche verso tutto ciò che non è chirale, come il punto di fusione, il punto di ebollizione, l’indice di rifrazione, lo spettro infrarosso, la solubilità in uno stesso solvente, o la velocità di reazione con i reagenti achirali.
Le differenze emergono quando la coppia enantiomerica viene fatta interagire con fenomeni chimici e fisici che abbiano natura chirale.
- Dal punto di vista chimico, due enantiomeri possono essere distinti quando si trovano a interagire con strutture chirali, come il sito di legame di un recettore chirale o il sito attivo di un enzima chirale.
- Dal punto di vista fisico due enantiomeri differiscono nelle interazioni con la luce polarizzata, che ha proprietà chirali, sono cioè dotati di attività ottica.[6]
Chiralità e attività ottica
L’attività ottica di materiali come il quarzo e, più importante, di composti organici come zuccheri o l’acido tartarico, fu scoperta nel 1815 dallo scienziato francese Jean-Baptiste Biot.[7]
Le molecole chirali possono essere classificate sulla base della direzione in cui viene ruotato il piano della luce polarizzata, rispetto al punto di vista dell’osservatore, quando attraversa una soluzione che le contenga.
- Se una soluzione di un enantiomero ruota il piano della luce polarizzata in senso orario, la molecola viene definita destrogira o destrorotatoria, dal latino dexter che significa “destro”, e suo nome è preceduto dai prefissi (+) o d–, da dextro-.
- Se una soluzione di un enantiomero ruota il piano della luce polarizzata in senso antiorario, la molecola è definita levogira o levorotatoria, dal latino laevus che significa “sinistro”, e il suo nome è preceduto dai prefissi (–) o l–, da laevo-.[8]
Ovviamente, considerando una coppia di enantiomeri, uno sarà destrogiro e l’altro non potrà che essere levogiro.
Al momento non è ancora possibile predire in modo affidabile ne la grandezza, la direzione, o il segno della rotazione del piano della luce polarizzata indotta da un enantiomero. D’altro canto, neppure l’attività ottica di una molecola da alcun tipo di informazione riguardo alla disposizione spaziale dei gruppi chimici legati al centro di chiralità.
Nota: un sistema contenente molecole che abbiamo lo stesso senso di chiralità è chiamato enantiomericamente puro o enantiopuro.[9]
Pasteur e la scoperta degli enantiomeri
Nel 1848, trentatre anni dopo il lavoro di Biot, studi sull’attività ottica delle molecole portarono Louis Pasteur, che di Biot era stato studente, a notare che, a seguito della ricristallizzazione di una soluzione acquosa concentrata di sodio ammonio tartrato (che era otticamente inattiva), di per se otticamente inattiva, precipitavano due tipi di cristalli che erano l’uno l’immagine speculare dell’altro e non sovrapponibili.[10]
Dopo averli separati con delle pinzette, Pasteur si accorse che le soluzioni ottenute sciogliendo quantità equimolari dei due cristalli erano otticamente attive e, cosa forse più interessante, l’angolo di rotazione del piano della luce polarizzata era lo stesso ma di segno opposto. Poiché queste differenze nell’attività ottica erano dovute alle molecole di sodio ammonio tartrato disciolte, Pasteur ipotizzò che le molecole stesse dovessero essere l’una l’immagine speculare dell’altra e non sovrapponibili, al pari dei loro cristalli, quelli che oggi chiamiamo enantiomeri.[11]
Fu Pasteur che per primo coniò il termine asimmetria per descrivere questa proprietà, definita in seguito chiralità da Lord Kelvin.[12]
Miscele racemiche
Una soluzione che contenga quantità equimolari di ciascun membro di una coppia di enantiomeri è detta miscela racemica o racemo. Queste soluzioni sono otticamente inattive in quanto l’effetto rotatorio di un enantiomero è esattamente compensato da quello dell’altro enantiomero.[13]
A differenza di quanto accade nei processi biochimici, la sintesi chimica di molecole chirali che non preveda il ricorso a reagenti chirali, o che non sia seguita da metodiche di separazione degli enantiomeri, porta inevitabilmente alla produzione di una miscela racemica.[9]
Il settore che più a risentito di questo fenomeno è quello della chimica farmaceutica. Come detto in precedenza, due enantiomeri sono molecole differenti. Molti farmaci chirali sono sintetizzati come miscele racemiche, ma molto spesso l’attività farmacologica desiderata è presente solamente in uno dei due enantiomeri, detto eutomero. L’altro enantiomero, detto distomero, è inattivo o meno attivo.[14]
Un esempio è il farmaco antiinfiammatorio ibuprofene, un derivato arilpropionico: solamente l’enantiomero S, così definito sulla base delle regola del sistema di nomenclatura detto sistema RS, è dotato di attività farmacologica.
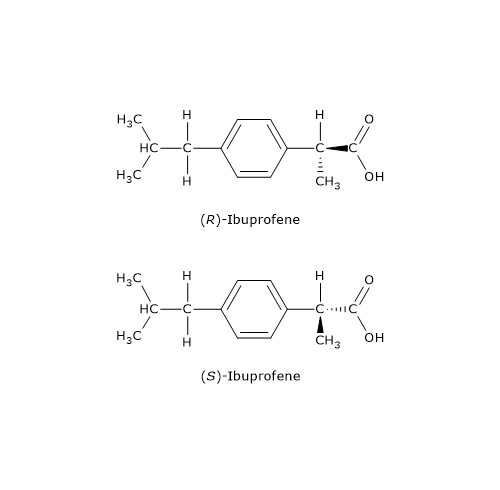
I derivati arilpropionici vengono venduti come miscele racemiche perché a livello epatico una racemasi converte il distomero in eutomero.[15]
Tuttavia è anche possibile che il distomero produca effetti dannosi e debba essere eliminato dalla miscela racemica. Un tragico esempio è la talidomide, un sedativo e anti-nausea commercializzato come miscela racemica dagli anni ‘50 fino al 1961, e utilizzato anche in gravidanza.
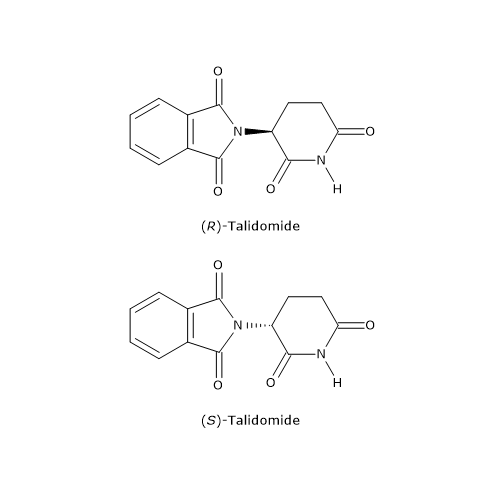
Il distomero, l’enantiomero S, poteva causare gravi difetti alla nascita, in particolare la focomelia (malformazione degli arti). Questo è forse l’esempio più eclatante dell’importanza delle proprietà chirali delle molecole, che ha poi spinto gli organismi di salute pubblica a promuovere, da parte dell’industria farmaceutica, la sintesi di farmaci, talidomide compresa, contenenti un singolo enantiomero.[16]
Centri di chiralità
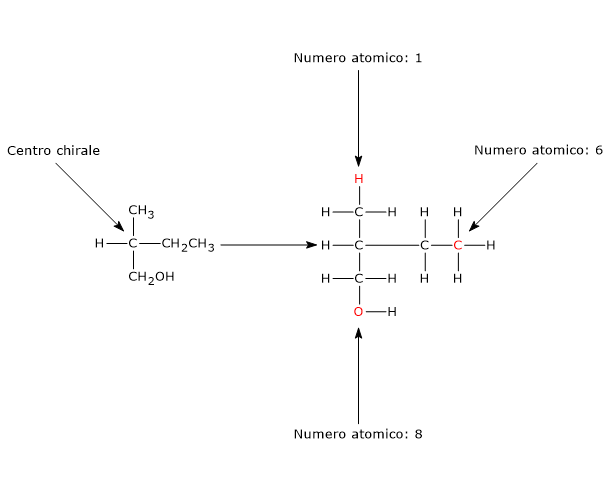
Un qualsiasi atomo tetraedrico che leghi quattro ligandi differenti può essere un centro chirale.
L’esempio classico è l’atomo di carbonio, ma anche altri atomi appartenenti al gruppo IVA della tavola periodica, come i semimetalli silicio e il germanio, formano composti a struttura tetraedrica e possono essere centri di chiralità.[2]
Anche l’atomo di fosforo negli organofosfati ha una geometria tetraedrica, quindi, quando lega quattro diversi sostituenti, è un centro chirale.[4]
L’atomo di azoto di un’ammina terziaria, un’ammina dove tre gruppi organici differenti sono legati all’azoto, è un centro chirale. In questi composti l’atomo di azoto è disposto al centro di un tetraedro e i suoi quattro orbitali ibridi sp3 sono diretti verso i vertici. Tre di questi sono occupati dai tre sostituenti, mentre verso il quarto è diretta la coppia di elettroni solitaria.
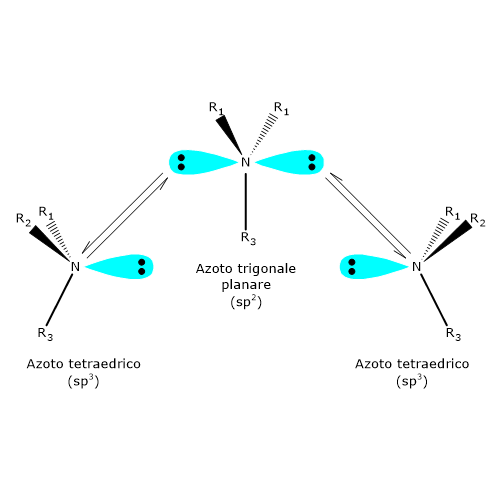
A temperatura ambiente, l’azoto inverte rapidamente la sua configurazione. Il fenomeno è noto come inversione dell’azoto, ossia, una rapida oscillazione dell’atomo e dei suoi ligandi, nel corso della quale l’azoto passa attraverso uno stato di transizione planare in cui ha ibridazione sp2. Come conseguenza, se l’azoto è il solo centro chirale della molecola, non c’è attività ottica in quanto si viene a formare una miscela racemica. L’inversione di configurazione viene evitata solamente in alcuni casi in cui l’azoto fa parte di una struttura ciclica che la impedisce. Dunque, la presenza di un centro chirale può non essere sufficiente per permettere la separazione dei rispettivi enantiomeri.[4]
Nota: nel 1874, Jacobus Henricus van ‘t Hoff e Joseph Achille Le Bel basandosi anche sui risultati ottenuti da Pasteur, per primi ipotizzarono la “teoria dell’atomo di carbonio tetraedrico”. Per questo lavoro van ’t Hoff ricevette il primo premio Nobel per la chimica nel 1901.[17]
Molecole chirali senza centri chirali
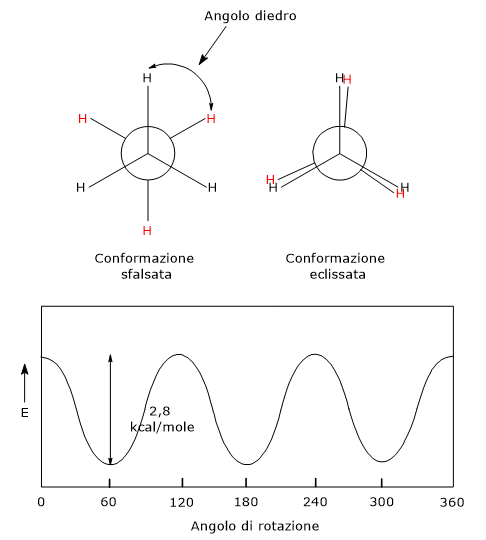
La chiralità può essere presente anche in assenza di un centro chirale, ed essere conseguenza della mancanza di libera rotazione attorno a un legame doppio o singolo.[18] Ciò ad esempio si osserva nel caso dei:
- derivati allenici, composti organici che presentano due doppi legami cumulati, cioè due doppi legami localizzati sullo stesso atomo di carbonio;[19]
- derivati bifenilici: composti costituiti da due anelli aromatici collegati da un singolo legame che non possono ruotare liberamente a causa dell’impedimento sterico dei sostituenti.[20]
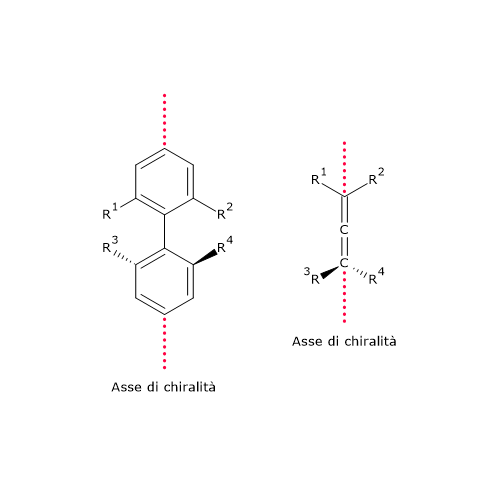
In questo caso la chiralità è conseguente alla presenza di un asse chirale piuttosto che di un centro chirale. Questo tipo di chiralità è noto come chiralità assiale.[21]
Composti meso
I composti meso o forme meso sono stereoisomeri che possiedono due o più centri chirali ma sono sovrapponibili alla loro immagine speculare, dunque sono achirali, e come tali otticamente inattivi. Inoltre posseggono un piano di simmetria interno che taglia la molecola in due metà, ognuna immagine speculare dell’altra.[8]
I composti meso sono classificati come diastereoisomeri, ossia stereoisomeri diversi dagli enantiomeri.
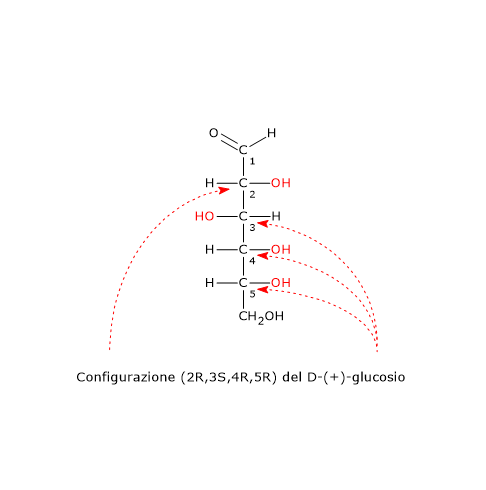
Per una molecola con n centri di chiralità il numero massimo di possibili stereoisomeri è 2n.[2]
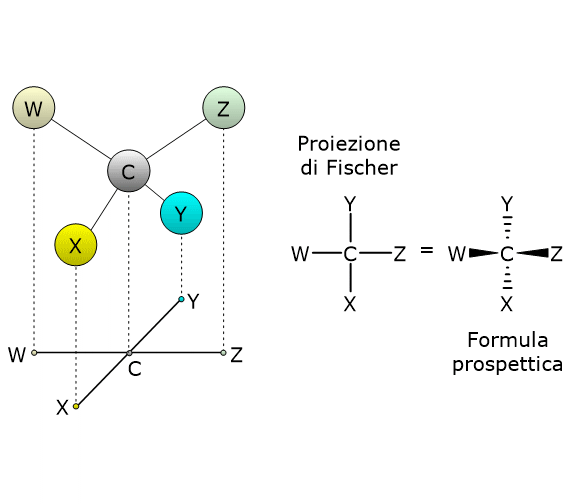
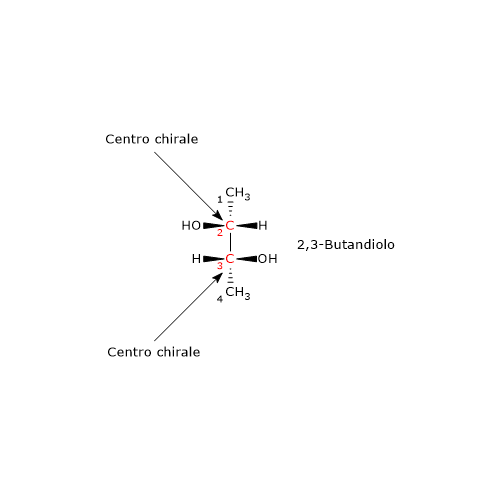
Si consideri il 2,3-butandiolo. La molecola presenta due centri di chiralità, i carboni 2 e 3, quindi si potrebbero avere 22 = 4 stereoisomeri, le cui strutture sono riportate in figura, secondo le proiezioni di Fischer, e indicate come A, B, C, D.
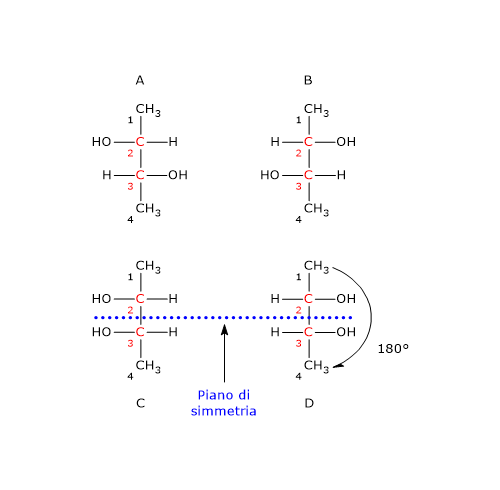
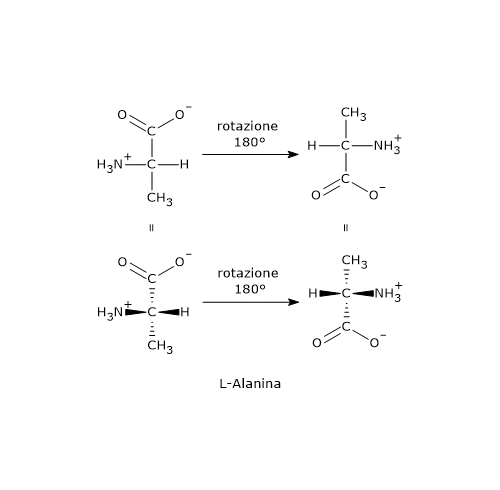
- Le strutture A e B sono l’una l’immagine speculare dell’altra e non sono sovrapponibili, dunque sono una coppia di enantiomeri.
- Le strutture C e D sono l’una l’immagine speculare dell’altra, ma sono sovrapponibili. Se infatti si ruota la struttura C o D di 180°, le due strutture si possono sovrapporre. Quindi non sono enantiomeri: sono la stessa molecola scritta con orientamento differente.
Inoltre hanno un piano di simmetria interno che le suddivide in due metà, l’una immagine speculare dell’altra. Dunque, la struttura C (o D) è un composto meso perché ha centri chiarali, è sovrapponibile alla sua immagine speculare e presenta un piano di simmetria interno che divide la molecola in due metà speculari.[2]
Bibliografia
- ^ Chirality, in IUPAC Compendium of Chemical Terminology, 5th ed. International Union of Pure and Applied Chemistry; 2025. Online version 5.0.0, 2025. doi:10.1351/goldbook.C01058
- ^ a b c d e Solomons T.W.G., Fryhle C.B., Snyder S.A. Solomons’ organic chemistry. 12th Edition. John Wiley & Sons Incorporated, 2017.
- ^ Kelvin W.T. Baltimore lectures on molecular dynamics and the wave theory of light. Clay C. J., London: 1904:619. https://archive.org/details/baltimorelecture00kelviala/mode/2
- ^ a b c Morsch L., Farmer S., Cunningham K., Sharrett Z., Shea K.M. Organic Chemistry II. Open Educational Resources: Textbooks, Smith College, Northampton, MA, 2023. https://scholarworks.smith.edu/textbooks/6
- ^ Enantiomer, in IUPAC Compendium of Chemical Terminology, 5th ed. International Union of Pure and Applied Chemistry; 2025. Online version 5.0.0, 2025. doi:10.1351/goldbook.E02069
- ^ Gogoi A., Konwer S., Zhuo G.Y. Polarimetric measurements of surface chirality based on linear and nonlinear light scattering. Front Chem 2021;8:611833. doi:10.3389/fchem.2020.611833
- ^ Flack H.D. Louis Pasteur’s discovery of molecular chirality and spontaneous resolution in 1848, together with a complete review of his crystallographic and chemical work. Acta Crystallogr A 2009;65(Pt 5):371-89. doi:10.1107/S0108767309024088
- ^ a b Soderberg T. Organic chemistry with a biological emphasis. Volume I. Chemistry Publications. 2019. https://digitalcommons.morris.umn.edu/chem_facpubs/1
- ^ a b Ariëns E.J. Stereochemistry: a source of problems in medicinal chemistry. Med Res Rev 1986;6(4):451-66. doi:10.1002/med.2610060404
- ^ Vantomme G., Crassous J. Pasteur and chirality: a story of how serendipity favors the prepared minds. Chirality 2021;33(10):597-601. doi:10.1002/chir.23349
- ^ Geison G.L. The private science of Louis Pasteur. Princeton University Press, 2014.
- ^ Pasteur L. Memoires sur la relation qui peut exister entre la forme crystalline et al composition chimique, et sur la cause de la polarization rotatoire. C R Acad Sci 1848;26:535‐538.
- ^ Racemate, in IUPAC Compendium of Chemical Terminology, 5th ed. International Union of Pure and Applied Chemistry; 2025. Online version 5.0.0, 2025. 10.1351/goldbook.R05025
- ^ Tamatam R., Shin D. Asymmetric synthesis of US-FDA approved drugs over five years (2016-2020): a recapitulation of chirality. Pharmaceuticals (Basel) 2023;16(3):339. doi:10.3390/ph16030339
- ^ Evans A.M., Nation R.L., Sansom L.N., Bochner F., Somogyi A.A. The relationship between the pharmacokinetics of ibuprofen enantiomers and the dose of racemic ibuprofen in humans. Biopharm Drug Dispos 1990;11(6):507-18. doi:10.1002/bdd.2510110605
- ^ Tokunaga E., Yamamoto T., Ito E., Shibata N. Understanding the thalidomide chirality in biological processes by the self-disproportionation of enantiomers. Sci Rep 2018;8(1):17131. doi:10.1038/s41598-018-35457-6
- ^ Jacobus H. van ‘t Hoff – Facts. NobelPrize.org. Nobel Prize Outreach 2025. Sun. 1 Jun 2025. https://www.nobelprize.org/prizes/chemistry/1901/hoff/facts/
- ^ Capozziello S., Lattanzi A. Geometrical approach to central molecular chirality: a chirality selection rule. Chirality 2003;15:227-230. doi:10.1002/chir.10191
- ^ Ōki M. The chemistry of rotational isomers. New York: Springer; 1993.
- ^ Runge W. The chemistry of the allenes. Vol. 2, Landor S R: Academic Press; 1982.
- ^ Axial chirality, in IUPAC Compendium of Chemical Terminology, 5th ed. International Union of Pure and Applied Chemistry; 2025. Online version 5.0.0, 2025. doi:10.1351/goldbook.A00547
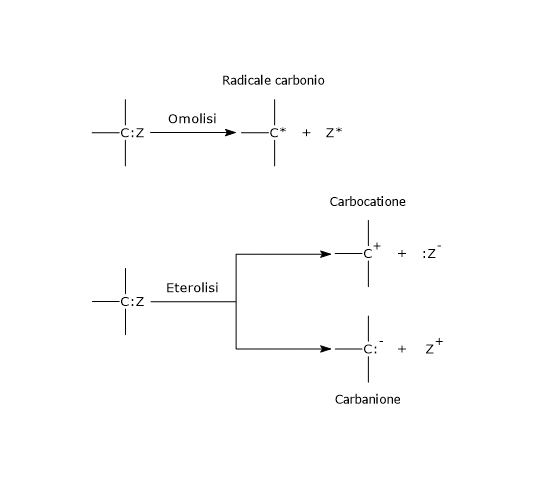 Nell’eterolisi, la rottura del legame covalente porta alla formazione di due atomi dotati di carica, ossia due ioni, un catione e un anione, poiché entrambi gli elettroni di legame vengono presi da uno dei due atomi precedentemente legati, quello più elettronegativo.[4] La reazione può procedere in due modi:
Nell’eterolisi, la rottura del legame covalente porta alla formazione di due atomi dotati di carica, ossia due ioni, un catione e un anione, poiché entrambi gli elettroni di legame vengono presi da uno dei due atomi precedentemente legati, quello più elettronegativo.[4] La reazione può procedere in due modi:
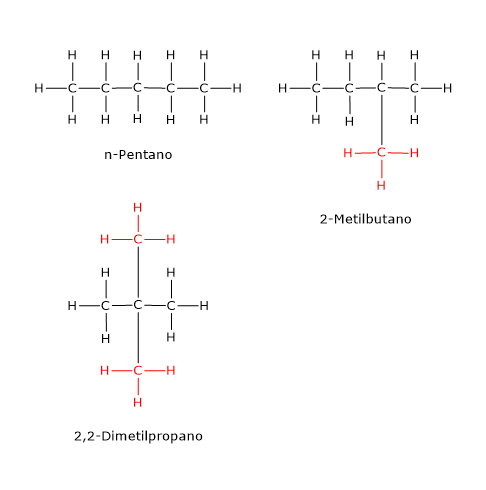 Esempi di isomeria di catena si trovano anche tra
Esempi di isomeria di catena si trovano anche tra