Il complesso della piruvato deidrogenasi (PDC) è un complesso multienzimatico mitocondriale composto da tre differenti enzimi:
- piruvato deidrogenasi (E1; EC 1.2.4.1);
- diidrolipoil transacetilasi (E2; EC 2.3.1.12);
- diidrolipoil deidrogenasi (E3; EC 1.8.1.4).
La stechiometria delle subunità e la dimensione del complesso variano a seconda della specie, con masse molecolari comprese tra 4 e 10 milioni di Dalton.[1]
Fanno parte del complesso multienzimatico anche cinque differenti coenzimi e, nelle piante, nei funghi e, tra gli animali, nei volatili e nei mammiferi, due enzimi con attività regolatoria: la piruvato deidrogenasi chinasi (EC 2.7.11.2) e la piruvato deidrogenasi fosfatasi (EC 3.1.3.43). Negli eucarioti, il complesso include una proteina di legame detta E3BP.[2]
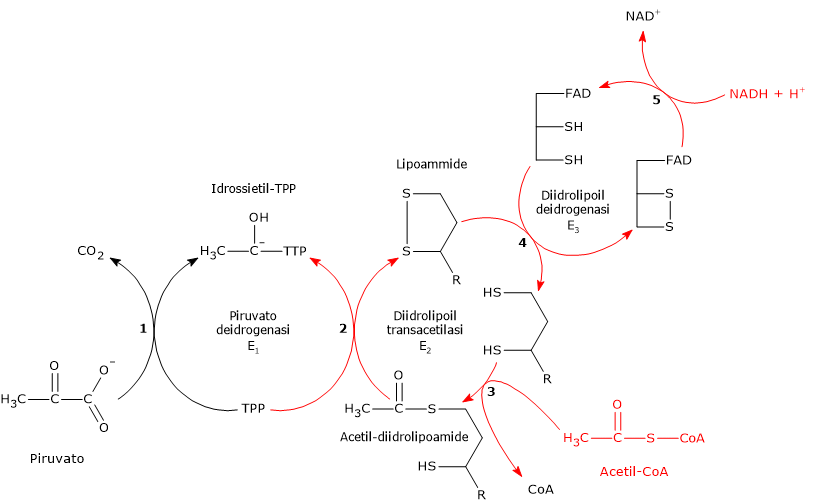
Il complesso della piruvato deidrogenasi catalizza, attraverso una sequenza di cinque reazioni, la decarbossilazione ossidativa del piruvato a dare il gruppo acetilico dell’acetil-coenzima A (acetil-CoA), una molecola di anidride carbonica (CO2), con il rilascio di due elettroni trasferiti al NAD+.[3]
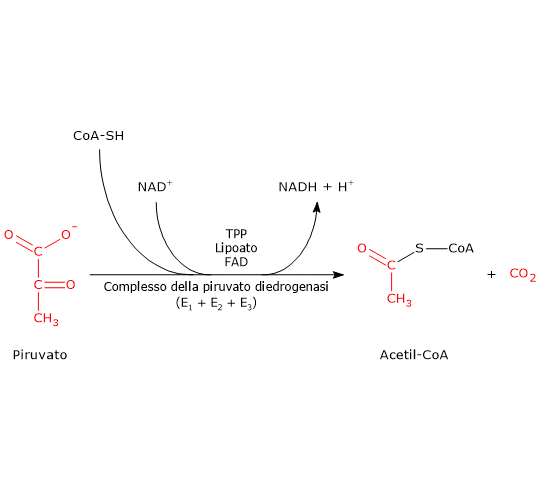
In condizioni fisiologiche, la reazione complessiva è essenzialmente irreversibile (ΔG°’ ≈ −8,0 kcal/mol; −33,4 kJ/mol), e richiede l’intervento sequenziale dei tre enzimi, le cui attività risultano coordinate.[4] Nel corso delle reazioni, i prodotti intermedi rimangono legati agli enzimi, e al termine della sequenza catalitica il complesso multienzimatico è pronto per il ciclo successivo.[5]
Nota: PDC catalizza le stesse reazioni attraverso meccanismi simili in tutti gli organismi in cui è presente.[6]
In Sintesi: Punti Chiave
- Funzione: catalizza la decarbossilazione ossidativa irreversibile del piruvato in acetil-CoA.
- Struttura: complesso multienzimatico mitocondriale (E1-E2-E3) che richiede cinque cofattori e due enzimi regolatori.
- Meccanismo e Regolazione: utilizza la canalizzazione del substrato ed è strettamente controllato da inibizioni allosteriche e modificazioni covalenti.
- Ruolo Metabolico: agisce da ponte tra la glicolisi e il ciclo dell’acido citrico.
Indice
- Coenzimi del complesso della piruvato deidrogenasi
- Localizzazione cellulare del complesso della piruvato deidrogenasi
- Funzioni del complesso della piruvato deidrogenasi
- Trasporto del piruvato nella matrice mitocondriale
- Struttura del complesso della piruvato deidrogenasi
- Struttura della piruvato deidrogenasi
- Struttura della diidrolipoil transacetilasi
- Struttura della diidrolipoil deidrogenasi
- Reazione della piruvato deidrogenasi
- Reazione della diidrolipoil transacetilasi
- Reazione della diidrolipoil deidrogenasi
- Regolazione dell’attività del complesso della piruvato deidrogenasi
- Bibliografia
Coenzimi del complesso della piruvato deidrogenasi
Cinque coenzimi partecipano alle reazioni catalizzate dal complesso della piruvato deidrogenasi: la tiamina pirofosfato (TPP), la flavina adenina dinucleotide (FAD), il coenzima A (CoA), la nicotinammide adenina dinucleotide (NAD) e l’acido lipoico.[1]
Tiamina pirofosfato
La tiamina pirofosfato deriva dalla tiamina o vitamina B1, di cui rappresenta la forma biologicamente attiva. TPP è il coenzima della piruvato deidrogenasi, cui è strettamente, ma non covalentemente, legata. Il suo ruolo è quello di trasportatore di gruppi idrossietilici o “aldeidici attivati”.[2]
Flavina adenina dinucleotide
La flavina adenina dinucleotide deriva dalla riboflavina o vitamina B2, di cui rappresenta una delle forme attive; l’altra è la flavina mononucleotide (FMN).
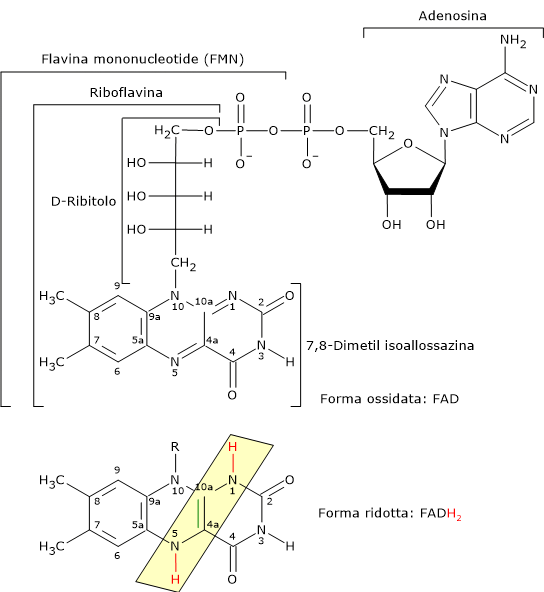
Il FAD è il coenzima della diidrolipoil deidrogenasi, cui è saldamente legato. Il suo ruolo, analogamente al NAD, è quello di trasportatore di elettroni, nello specifico due elettroni e due protoni (2H+ + 2e−).[5]
Coenzima A
Il coenzima A è formato da una β-mercaptoetilamina legata attraverso legame ammidico all’acido pantotenico o vitamina B5. A sua volta la vitamina è unita, attraverso un ponte pirofosfato, a una 3′-fosfoadenosina.
Il CoA partecipa alla reazione catalizzata dalla diidrolipoil transacetilasi. Il suo ruolo è quello di trasportatore di gruppi acilici.[7]
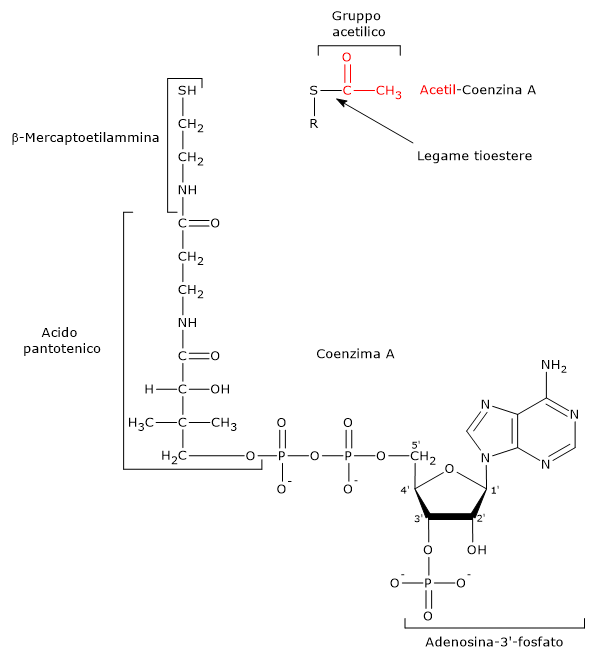
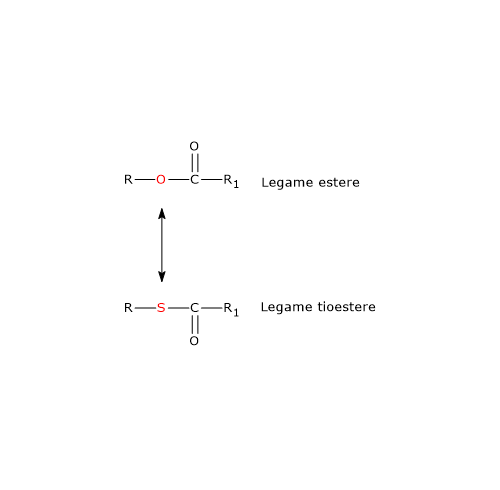
La porzione del coenzima A corrispondente alla β-mercaptoetilamina termina con un gruppo sulfidrilico (–SH). Si tratta di un tiolo reattivo, cruciale per il ruolo svolto dal coenzima in quanto i gruppi acilici trasportati, legati attraverso un legame tioestere, hanno un’elevata energia libera standard di idrolisi. Ciò fornisce loro un elevato potenziale di trasferimento, pari a −7,5 kcal/mol (−31,5 kJ/mol). Degno di nota che questo potenziale di trasferimento è più esoergonico, anche se per una differenza di soli +0,2 kcal/mol (+1 kJ/mol), rispetto a quello per l’idrolisi dell’ATP ad ADP e Pi.
Pertanto, i tioesteri hanno un elevato potenziale di trasferimento del gruppo acilico e sono in grado di donarlo a numerose molecole accettrici. In altre parole il gruppo acilico legato al coenzima A può essere considerato come una forma attivata pronta per il trasferimento. È anche possibile affermare che la formazione del legame tioestere permette di conservare parte dell’energia metabolica derivante dall’ossidazione del carburante metabolico. E va sottolineato che il coenzima A è anche abbreviato come CoA-SH, proprio per enfatizzare il ruolo svolto dal gruppo tiolico.[4]
Nota: nel legame tioestere è presente un atomo di zolfo nella posizione in cui nel legame estere è presente un atomo di ossigeno.[8]
Nicotinammide adenina dinucleotide
La nicotinammide adenina dinucleotide (NAD+) può essere prodotta a partire dal triptofano, un amminoacido essenziale, o dalla niacina o vitamina B3 o vitamina PP (da Pellagra-Preventing), la fonte della parte nicotinammidica.
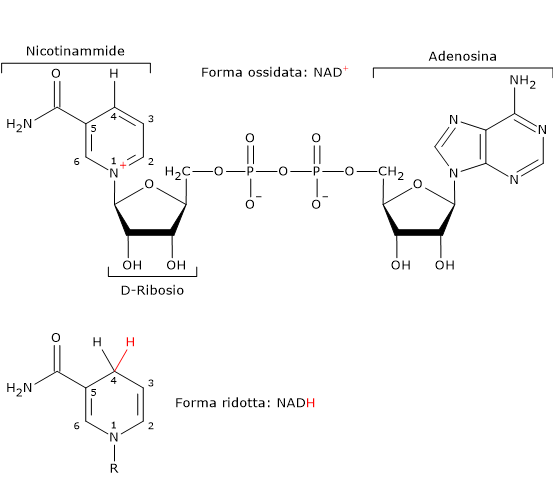
Il NAD+ partecipa alla reazione catalizzata dalla diidrolipoil deidrogenasi. Il suo ruolo, analogamente al FAD, è quello di trasportatore di elettroni, o ioni idruro.[4]
Acido lipoico
A differenza degli altri coenzimi del complesso della piruvato deidrogenasi, l’acido lipoico non deriva, direttamente o indirettamente, da vitamine e/o amminoacidi essenziali, ossia da precursori che non possono essere sintetizzati de novo dall’organismo e devono essere assunti con la dieta.
L’acido lipoico è il coenzima della diidrolipoil transacetilasi. Il coenzima è legato all’enzima a mezzo di un legame ammidico con l’ε-ammino gruppo di un residuo di lisina a formare un residuo di lipoil-lisina o lipoammide. Il coenzima accoppia il trasferimento di elettroni con quello di gruppi acilici.[9]
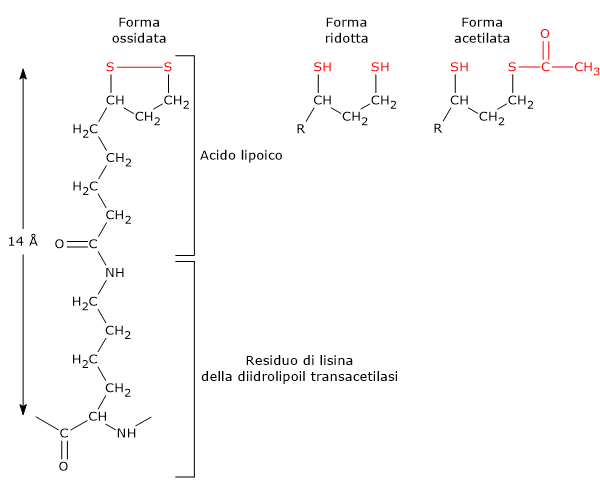
L’acido lipoico possiede due gruppi tiolici che possono andare incontro a ossidazione intramolecolare reversibile a dare un ponte disolfuro (–S–S–), similmente a quanto può accadere tra due residui di cisteina (Cys) di una proteina.
Poiché il ponte disolfuro (un disolfuro ciclico) può andare incontro a reazioni di ossidoriduzione, nel corso delle reazioni catalizzate dal complesso è dapprima ridotto a diidrolipoammide, un ditiolo (la forma ridotta del gruppo prostetico), e quindi riossidato nel disolfuro ciclico.[1]
Nota
Molti enzimi necessitano per la loro attività catalitica di piccoli componenti non proteici chiamati cofattori. I cofattori possono essere ioni metallici o piccole molecole organiche o metallo-organiche e sono classificati come coenzimi e gruppi prostetici.
Un gruppo prostetico è un cofattore che si lega in modo permanente alla proteina, a mezzo di un legame non covalente o covalente. TPP, FAD e acido lipoico sono considerati gruppi prostetici del complesso della piruvato deidrogenasi a causa del loro legame stretto.
Un coenzima, ad esempio il CoA o il NAD+, è un cofattore che non è legato in modo permanente all’enzima.[5]
Localizzazione cellulare del complesso della piruvato deidrogenasi
Negli eucarioti, il complesso della piruvato deidrogenasi, al pari degli enzimi del ciclo dell’acido citrico e di quelli per l’ossidazione degli acidi grassi, si trova nel mitocondrio, associato alla superficie della membrana mitocondriale interna che guarda la matrice.
Nei procarioti si trova nel citosol.[6]
Funzioni del complesso della piruvato deidrogenasi
Il complesso della piruvato deidrogenasi ha essenzialmente due funzioni: produrre acetil-CoA e NADH.
Il gruppo acetilico legato al coenzima A, un acetato attivato, può andare incontro a due differenti destini.
- Può essere ossidato a due molecole di anidride carbonica attraverso le reazioni del ciclo dell’acido citrico, consentendo la “raccolta” di una parte dell’energia potenziale sotto forma di ATP o GTP.
- Può essere utilizzato per la sintesi di acidi grassi, colesterolo, steroidi, isoprenoidi, corpi chetonici e acetilcolina.
È quindi possibile affermare che, a seconda delle condizioni metaboliche e/o del tipo di cellula, il complesso della piruvato deidrogenasi indirizza intermedi carboniosi derivanti dal catabolismo degli amminoacidi e del glucosio verso:
- il ciclo dell’acido citrico e quindi la produzione di energia, come nel muscolo scheletrico in condizioni aerobiche e nel muscolo cardiaco;
- la sintesi dei lipidi e dell’acetilcolina.[10]
Negli organismi aerobi, il NADH può essere ossidato a NAD+ mediante la cessione di uno ione idruro al Complesso I della catena di trasporto degli elettroni mitocondriale. Quest’ultima trasferisce i due elettroni all’ossigeno molecolare (O2), permettendo la produzione di 2,5 molecole di ATP per coppia di elettroni.[1]
Nota: negli organismi anaerobi sono presenti accettori di elettroni alternativi all’ossigeno, come nitrati o solfati.[11]
Dal punto di vista concettuale, il complesso della piruvato deidrogenasi rappresenta il ponte di collegamento tra glicolisi e ciclo dell’acido citrico. Tuttavia, vista l’irreversibilità della reazione complessiva catalizzata dal complesso, si tratta di un ponte a senso unico. Il piruvato può essere decarbossilato e ossidato, con formazione di acetil-CoA, mentre non è possibile il percorso inverso, ossia la conversione dell’acetil-CoA in piruvato.
L’irreversibilità della reazione, insieme all’assenza di vie metaboliche alternative, spiega perché non è possibile utilizzare l’acetil-CoA, e quindi gli acidi grassi, per la gluconeogenesi.[5]
Altre fonti di acetil-CoA
Il gruppo acetilico dell’acetil-CoA può derivare, oltre che dal piruvato, dall’ossidazione degli acidi grassi e dal catabolismo di molti amminoacidi. Tuttavia, a prescindere dalla sua origine, l’acetil-CoA rappresenta un veicolo di ingresso di nuove unità carboniose nel ciclo dell’acido citrico. È anche possibile affermare che il gruppo acetilico dell’acetil-CoA rappresenta la forma in cui la maggior parte del carbonio entra nel ciclo.[10]
Trasporto del piruvato nella matrice mitocondriale
Negli eucarioti, la glicolisi avviene nel citosol, mentre tutti i passaggi successivi del metabolismo aerobico, quindi le reazioni catalizzate dal complesso della piruvato deidrogenasi, il ciclo dell’acido citrico, la catena di trasporto degli elettroni e la fosforilazione ossidativa, si svolgono nel mitocondrio.[12]
Similmente a quanto accade per la maggior parte degli altri anioni e metaboliti, il passaggio del piruvato attraverso la membrana mitocondriale esterna è probabilmente mediato da un canale anionico voltaggio-dipendente relativamente non specifico. Di contro, il passaggio attraverso la membrana mitocondriale interna è assicurato da uno specifico trasportatore formato da due proteine denominate MPC1 e MPC2 (Mitochondrial Pyruvate Carrier) che vanno a formare un complesso etero-oligomerico nella membrana, catalizzando un simporto piruvato/H+ verso la matrice.[13]
Struttura del complesso della piruvato deidrogenasi
Sebbene il complesso della piruvato deidrogenasi sia formato da copie multiple di tre enzimi differenti e catalizzi le medesime reazioni tramite meccanismi simili in tutti gli organismi in cui è presente, ha una struttura quaternaria molto varia.[6]
Struttura del complesso di E. coli
Il primo complesso di cui fu analizzata la struttura fu quello di E. coli, grazie al lavoro di Lester Reed. Nel complesso multienzimatico, che ha un peso di circa 4600 kDa e un diametro di circa 300 Å, 24 unità di diidrolipoil transacetilasi vanno a formare una struttura con simmetria cubica, ossia, gli enzimi associati in trimeri sono disposti agli angoli del cubo. Dimeri di piruvato deidrogenasi si associano al nucleo di diidrolipoil transacetilasi, al centro di ognuno dei 12 bordi del cubo, per un totale di 24 unità. Infine, dimeri di diidrolipoil deidrogenasi si vanno a disporre al centro di ognuna delle sei facce del cubo, per un totale di 12 unità. Si noti che l’intero complesso è costituito da 60 unità. Anche nella maggior parte degli altri batteri Gram-negativi si osserva un’analoga struttura con simmetria cubica.[14]
Struttura del complesso negli eucarioti e nei batteri Gram-positivi
In alcuni batteri Gram-positivi e negli eucarioti il complesso della piruvato deidrogenasi presenta una struttura dodecaedrica, ossia quella di un poliedro regolare con 20 vertici, 12 facce pentagonali e 30 bordi, con simmetria icosaedrica, anche detta simmetria I. Se si considera ad esempio il complesso presente nei mitocondri, questo è il più grande complesso multienzimatico conosciuto. Ha un peso di circa 10000 kDa (10 MDa) e un diametro di circa 500 Å, dunque ben 5 volte le dimensioni di un intero ribosoma. Inoltre è abbastanza grande da poter essere visualizzato con il microscopio elettronico.[15][16]
Il complesso è costituito da un nucleo dodecaedrico avente un diametro di circa 25 nm e formato, come nei batteri Gram-negativi, dalla diidrolipoil transacetilasi, ma composto da 20 trimeri dell’enzima, per un totale di 60 unità, disposti ai vertici della struttura. Il nucleo è circondato da 30 unità di piruvato deidrogenasi, una centrata su ogni bordo, e 12 unità di diidrolipoil deidrogenasi, una centrata su ogni faccia. L’intero complesso è quindi costituito da 102 unità.[2]
Subunità addizionali
La struttura quaternaria del complesso della piruvato deidrogenasi è ulteriormente complicata dalla presenza di tre subunità addizionali: la piruvato deidrogenasi chinasi, la piruvato deidrogenasi fosfatasi e la E3BP (E3-binding protein). La chinasi e la fosfatasi sono legate al nucleo di diidrolipoil transacetilasi.[4]
E3BP è legata a ognuna delle 12 facce pentagonali, e dunque presente in circa dodici copie. La proteina è necessaria per il legame della diidrolipoil deidrogenasi al nucleo formato dalla diidrolipoil transacetilasi, come dimostrato dal fatto che la sua proteolisi parziale riduce la capacità di legame della deidrogenasi.[17]
Nella E3-binding protein è possibile individuare un dominio C-terminale, privo di attività catalitica, e un dominio che contiene una lipoil-lisina, simile a quella della diidrolipoil transacetilasi, in grado anche di accettare un gruppo acetilico. Tuttavia, la rimozione di questo dominio non comporta alcuna riduzione dell’attività catalitica del complesso multienzimatico.[18]
Struttura della piruvato deidrogenasi
La piruvato deidrogenasi degli eucarioti e di alcuni batteri Gram-positivi è composta da due differenti catene polipeptidiche, indicate come α e β, disposte a formare un eterotetramero α2β2 simmetrico. Di contro, in E. coli e in altri batteri Gram-negativi le subunità sono fuse a formare un’unica catena polipeptidica e l’enzima risulta un omodimero.[6][19]
L’enzima presenta due siti attivi.
Considerando la struttura eterotetramerica della piruvato deidrogenasi di Bacillus (Geobacillus) stearothermophilus, un batterio Gram-positivo, ogni tiamina pirofosfato si lega tra i domini N-terminali di una subunità α e di una β, al termine di un canale a forma di imbuto profondo circa 21 Å che porta al sito attivo, con l’anello tiazolico, il suo gruppo reattivo, posizionato vicino all’ingresso del canale stesso.
All’ingresso del canale sono inoltre presenti due anse peptidiche conservate, essenziali sia per l’attività catalitica dell’enzima che per la sua regolazione.[20]
Architettura e struttura del sito attivo
L’analisi ai raggi X dell’enzima di B. stearothermophilus, quando lega sia TPP che il dominio centrale di legame (PSBD; peripheral subunit-binding domain) della diidrolipoil transacetilasi, che si lega al dominio C-terminale delle subunità β, ha evidenziato, oltre a un eterotetramero con una struttura estremamente compatta, che i due siti attivi hanno una struttura differente, in particolare riguardo alla disposizione delle due anse peptidiche conservate.[16] Infatti, in una subunità dell’enzima, in presenza della forma attivata della tiamina pirofosfato l’ansa più interna è disposta in modo da bloccare l’ingresso al sito attivo, mentre l’ansa all’imboccatura dell’altro sito attivo ha una conformazione tale da non bloccarne l’ingresso.[21]
Questo spiega, dal punto di vista strutturale, le differenze osservate nella velocità di legame del substrato esibite dai due siti attivi. Una disposizione e asimmetria simili sono state osservate anche in tutti gli altri enzimi tiamina pirofosfato–dipendenti di cui sia nota la struttura.
Oltre alla tiamina pirofosfato e a uno ione magnesio, localizzati in ciascuno dei due siti attivi dell’enzima, un terzo Mg2+ è posizionato al centro del tetramero, all’interno di un tunnel lungo circa 20 Å, riempito di solvente, che collega i due siti attivi. Il tunnel è in gran parte delimitato da 10 residui amminoacidici conservati, rispettivamente sei residui di glutammato (Glu) e quattro di aspartato (Asp), che provengono da tutte e quattro le subunità, più altri residui acidi attorno all’anello amminopirimidinico della TPP. Va invece sottolineata l’assenza di residui basici che neutralizzino i precedenti. Tunnel simili sono stati osservati in tutti gli enzimi tiamina pirofosfato dipendenti con struttura dimerica o tetramerica di cui sia caratterizzata la struttura cristallina. Un esempio è la transchetolasi, enzima della via del pentoso fosfato.[2]
Nota: B. stearothermophilus, membro del phylum Firmicutes, è stato da poco rinominato Geobacillus stearothermophilus.
Funzione del tunnel acido
Attraverso esperimenti di mutagenesi condotti sulla piruvato deidrogenasi di B. stearothermophilus è stato dimostrato che il tunnel ha un ruolo nel meccanismo catalitico.[22][23]
La sostituzione di alcuni dei suddetti residui acidi con amminoacidi neutri non altera, rispetto alla forma wild-type:
- l’efficienza dell’incorporazione dell’enzima modificato nel complesso multienzimatico;
- la struttura dei siti attivi;
- la struttura quaternaria dell’enzima.[24]
Tuttavia, la velocità di decarbossilazione risultava ridotta di oltre il 70% rispetto all’enzima wild-type; allo stesso modo, una volta che l’enzima mutante era inserito nel complesso della piruvato deidrogenasi, l’attività del complesso stesso risultava ridotta di oltre l’85%, sempre in confronto alla forma wild-type.[23] Ma a che cosa sono dovute queste riduzioni dell’efficienza catalitica?
Poiché la distanza tra gli amminoacidi sostituiti e i siti attivi dell’enzima è di 7 o più Å, questi amminoacidi sono troppo lontani dai siti attivi e non possono influenzare direttamente l’attività. È stato quindi proposto il meccanismo di seguito descritto.[3]
Meccanismo del filo protonico e comunicazione tra i siti attivi
Considerando l’apoenzima, TPP si lega rapidamente e con elevata affinità al primo sito attivo, viene attivata e il sito attivo si chiude. In questo modo lo zwitterione tiazolico risulta protetto dall’ambiente esterno.
Nel secondo sito attivo, invece, TPP si lega ma non viene attivata e il sito attivo rimane in conformazione aperta.[4]
Nel primo sito attivo, il piruvato reagisce con il C-2 tiazolico e la tiamina pirofosfato del secondo sito attivo, che agisce da acido generale, dona un protone al primo sito. Il risultato è la decarbossilazione del piruvato nel primo sito attivo e l’attivazione del coenzima nel secondo, che quindi viene chiuso.[3]
Si noti che, mentre l’attivazione della prima TPP è una conseguenza del legame al sito attivo, l’attivazione della seconda, e quindi del secondo sito attivo, è accoppiata alla decarbossilazione del piruvato nel primo sito attivo. In altri termini, mentre un sito attivo richiede la presenza di un acido generale, l’altro richiede una base generale.[2]
I protoni sono essenziali per l’attività catalitica e il loro trasferimento tra i siti attivi avviene attraverso il tunnel acido. Si tratta di un trasporto reversibile lungo una catena di gruppi accettori–donatori costituiti da residui di glutammato e aspartato e da molecole d’acqua presenti nel tunnel, che nell’insieme agiscono come un vero e proprio filo protonico (proton wire).
A differenza di molti altri enzimi, nei quali la comunicazione tra i siti attivi avviene tramite modificazioni conformazionali e riarrangiamenti delle subunità, nella piruvato deidrogenasi e negli altri enzimi TPP–dipendenti il proton wire rappresenta la base molecolare di tale comunicazione.[22]
A questo punto, l’oloenzima è formato e i siti attivi si trovano in equilibrio dinamico, passando vicendevolmente dallo stato dormiente a quello attivato (meccanismo flip-flop). Questo sembra essere lo stato fisiologico dell’enzima all’inizio di ogni ciclo catalitico.[19]
Implicazioni dell’asimmetria mediata dal filo protonico per la catalisi
Una conseguenza di un tale meccanismo è che, a mano a mano che i cicli catalitici si susseguono, i due siti attivi sono fuori fase tra di loro, ossia, mentre uno richiede una base generale, l’altro necessita di un acido generale, e viceversa.[22]
Da notare infine che un tale meccanismo permette anche il cambio di conformazione delle anse che chiudono i siti attivi, in modo da:
- coordinare l’assunzione dei substrati e il rilascio dei prodotti;
- spiegare l’asimmetria esistente tra i due siti attivi.[23]
Nota: un apoenzima è un enzima privo dei suoi cofattori, mentre un oloenzima è un apoenzima che lega i suoi cofattori. L’apoenzima è cataliticamente inattivo, mentre l’oloenzima è cataliticamente attivo.[5]
Struttura della diidrolipoil transacetilasi
Nella struttura della diidrolipoil transacetilasi è possibile individuare tre domini funzionalmente distinti: un dominio lipoilico N-terminale, un dominio centrale di legame, e un dominio catalitico C-terminale o acetiltransferasico. Questi domini sono connessi da sequenze di 20–40 residui amminoacidici ricche in alanina e prolina, amminoacidi idrofobici che sono inframezzati da residui dotati di carica. Queste sequenze di collegamento sono altamente flessibili ed estese, il che permette di tenere lontani gli uni dagli altri i tre domini.[25][26]
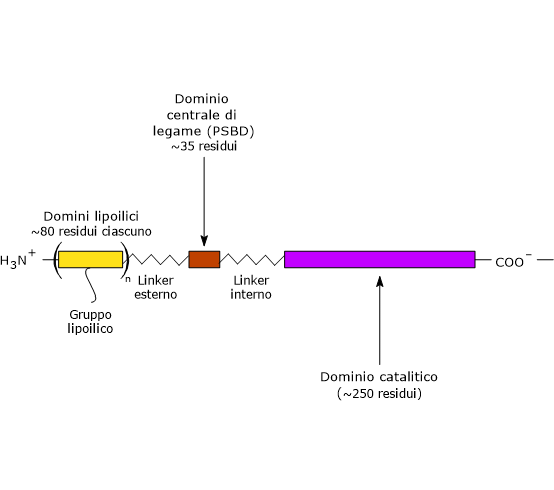
Nota: analoghe sequenze di collegamento flessibili sono presenti anche in E3BP.
Dominio lipoilico
Il dominio lipoilico N-terminale è composto da circa 80 residui amminoacidici ed è così chiamato perché lega l’acido lipoico. Il numero di questi domini varia in funzione della specie, da uno a tre. Ad esempio, è presente con una sola copia in Enterococcus faecalis, con due nei mammiferi e con tre in Azotobacter vinelandii ed E. coli.[27][28]
Il legame tra l’ɛ-ammino gruppo della catena laterale di una lisina e l’acido lipoico porta alla formazione di un braccio flessibile, la lipoil-lisina, che alla sua massima estensione ha una lunghezza di circa 14 Å. Se a questo si aggiunge la sequenza che collega il dominio N-terminale al dominio adiacente, la cui lunghezza è superiore ai 140 Å, si ottiene un braccio flessibile in grado di far oscillare il gruppo lipoilico tra i siti attivi della piruvato deidrogenasi e della diidrolipoil deidrogenasi, nonché di interagire con le unità di diidrolipoil transacetilasi vicine.[2][29]
Si noti che il numero di queste braccia flessibili in E. coli è pari a 3 x 24 = 72, mentre nei mammiferi a 2 x 60 = 120, sulla base del numero dei domini N-terminali e delle unità di diidrolipoil transacetilasi.
Da tutto ciò consegue che una piruvato deidrogenasi può acetilare numerose diidrolipoil transacetilasi, e una diidrolipoil deidrogenasi può riossidare diversi gruppi diidrolipoammidici.
Inoltre, si verificano anche:
- un interscambio di gruppi acetilici tra i gruppi lipoilici del nucleo di diidrolipoil transacetilasi;
- lo scambio sia di gruppi acetilici che di disolfuri tra le braccia flessibili.[25]
Dominio centrale di legame
PSBD è composto da circa 35 residui amminoacidici disposti a formare una struttura globulare che si lega sia alla piruvato deidrogenasi che alla diidrolipoil deidrogenasi, ossia aiuta a tenere unito il complesso multienzimatico.[30]
Dominio catalitico C-terminale
Il dominio catalitico C-terminale, che contiene il sito attivo, è composto da circa 250 residui amminoacidici disposti a formare una struttura simile a una gabbia vuota che contiene canali sufficientemente larghi da permettere ai substrati e ai prodotti di diffondere fuori e dentro. Ad esempio, la lipoammide e il coenzima A, i due substrati della diidrolipoil transacetilasi, si legano in conformazione estesa alle estremità opposte di un canale che si trova localizzato all’interfaccia tra ogni paio di subunità di ciascun trimero.[26][31]
Struttura della diidrolipoil deidrogenasi
La struttura della diidrolipoil deidrogenasi venne dedotta dallo studio dell’enzima in numerosi microrganismi. Presenta una struttura omodimerica, dove ciascuna catena polipeptidica, costituita da circa 470 amminoacidi, è ripiegata a dare quattro domini, dall’estremità N-terminale a quella C-terminale: un dominio legante il FAD, uno legante il NAD+, uno centrale e uno di interfaccia. Tutti i domini contribuiscono alla formazione del sito attivo.[32]
Il FAD si trova quasi completamente all’interno della proteina. Ciò è dovuto al fatto che, a differenza del NADH o dei tioli, è facilmente ossidabile e deve essere protetto dal solvente, in particolare dall’ossigeno. In assenza del NAD+, la catena fenolica di una tirosina (Tyr) altamente conservata, ad esempio la Tyr181 nel batterio Gram-negativo Pseudomonas putida, occlude la tasca di legame della nicotinammide, impedendo il contatto del FADH2 con il solvente.[2]
Quando invece il NAD+ è nel sito attivo, la catena fenolica della tirosina si dispone tra l’anello nicotinammidico e quello flavinico.[33]
Nel sito attivo della forma ossidata dell’enzima è presente un ponte disolfuro redox-attivo formato tra due residui di cisteina localizzati in un segmento altamente conservato della catena polipeptidica, ad esempio Cys43 e Cys48 in P. putida. Il ponte disolfuro è posizionato sul lato dell’anello flavinico opposto rispetto a quello rivolto verso l’anello nicotinamidico, e collega due giri consecutivi di un segmento di un’α-elica distorta; in assenza della distorsione, i carboni α delle due cisteine risulterebbero troppo distanti per consentirne la formazione.[34]
La diidrolipoil deidrogenasi possiede quindi due accettori di elettroni: il FAD e il ponte disolfuro redox-attivo.
Nota: gli anelli eterociclici del NAD e del FAD sono paralleli e interagiscono tramite forze di van der Waals; anche lo zolfo della Cys48 è in contatto di van der Waals con l’anello flavinico, sul lato opposto rispetto all’anello nicotinammidico.[2]
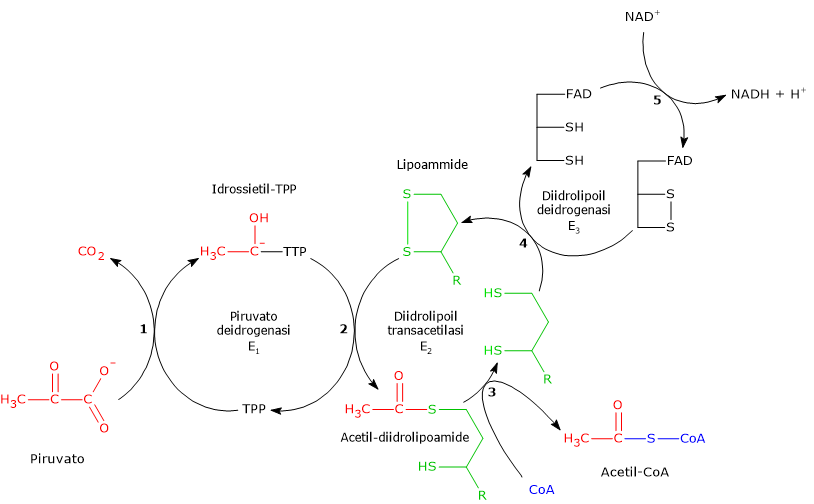
Reazione della piruvato deidrogenasi
La piruvato deidrogenasi, nella sequenza delle cinque reazioni catalizzate dai componenti del complesso della piruvato deidrogenasi, ne catalizza le prime due, ovvero:
- la decarbossilazione del piruvato a dare CO2 e l’intermedio idrossietil-TPP;
- l’acetilazione riduttiva del gruppo lipoilico della diidrolipoil transacetilasi.
La prima reazione è essenzialmente identica alla reazione catalizzata dalla piruvato decarbossilasi (EC 4.1.1.1), che opera una decarbossilazione non ossidativa nel corso della fermentazione del glucosio a etanolo. Quello che differisce è il destino del gruppo idrossietilico legato alla tiamina pirofosfato: nella reazione catalizzata dalla piruvato deidrogenasi viene trasferito all’enzima successivo della sequenza, la diidrolipoil transacetilasi, mentre nella reazione catalizzata dalla piruvato decarbossilasi è convertito in acetaldeide.[4]
Meccanismo di reazione: prima fase
Nelle reazioni catalizzate da enzimi TPP-dipendenti, l’anello tiazolico rappresenta la parte attiva, ma solo in forma di carbanione dipolare o ylide, ossia uno ione dipolare o zwitterione, con carica positiva sull’N-3 e carica negativa sul C-2. Di contro, l’anello tiazolico con carica positiva, ossia una carica positiva sull’azoto e nessuna carica sul C-2, può essere definito come la forma “dormiente” o inattiva.[35]
La reazione ha inizio con l’attacco nucleofilo da parte del carbanione sul C-2 al carbonio carbonilico del piruvato, che ha lo stato di ossidazione di un’aldeide. L’attacco porta alla formazione di un legame covalente tra il coenzima e il piruvato.
Segue la scissione del legame C-1–C-2 del piruvato. Questo porta al distacco del gruppo carbossilico, ossia del C-1 in forma di CO2, mentre i due restanti atomi di carbonio, il C-2 e il C-3, rimangono legati alla tiamina pirofosfato in forma di gruppo idrossietilico. La scissione del legame C-1–C-2, e quindi la decarbossilazione del piruvato, è favorita dal fatto che la carica negativa sul C-2, che è instabile, viene stabilizzata grazie alla presenza nell’anello tiazolico dell’azoto imminico carico positivamente, N-3, (C=N+), ossia grazie alla presenza di una struttura elettrofila o elettron-deficiente che funge da pozzo o trappola per gli elettroni, nella quale gli elettroni dei carbanioni possono delocalizzarsi per risonanza.[36]
A questo punto, l’intermedio stabilizzato per risonanza può essere protonato a dare idrossietil–TPP.[5]
Nota: questo primo passaggio della reazione catalizzata dalla piruvato deidrogenasi è quello in cui il complesso della piruvato deidrogenasi esercita la sua specificità di substrato. Inoltre, è anche il più lento dell’intera sequenza, ossia è quello che limita la velocità della reazione complessiva.[1]

Meccanismo di reazione: seconda fase
Di seguito, l’enzima catalizza l’ossidazione del gruppo idrossietilico a gruppo acetilico e il suo trasferimento sul braccio lipoammidico della diidrolipoil transacetilasi. La reazione ha inizio con la formazione di un carbanione sul carbonio idrossilico della idrossietil–TPP, a seguito della rimozione del protone legato al carbonio stesso da parte di una base dell’enzima.[5]
Segue l’attacco nucleofilo del carbanione al disolfuro della lipoammide, con formazione di un legame acetil-tioestere ad alta energia con uno dei due tioli. In questa reazione l’ossidazione del gruppo idrossietilico a gruppo acetilico è accompagnata dalla concomitante riduzione del ponte disolfuro della lipoammide: i due elettroni rimossi dal gruppo idrossietilico sono utilizzati per ridurre il ponte disolfuro. Questa reazione è quindi un’acetilazione riduttiva cui si accoppia la rigenerazione della forma attiva della piruvato deidrogenasi, ossia l’enzima con il C-2 dell’anello tiazolico nella forma deprotonata, la forma ilide o carbanione dipolare.[36]
Si noti che l’energia derivante dall’ossidazione del gruppo idrossietilico a gruppo acetilico permette la formazione del legame tioestere tra il gruppo acetilico e la lipoammide.[4]
Nota: il braccio di lipoil-lisina, presente in conformazione estesa, può accedere al sito attivo della piruvato deidrogenasi, dove è legata la TPP, permettendo il trasferimento dell’unità acetilica, generata per ossidazione dell’idrossietil–TPP, alla lipoammide stessa. Successivamente, il braccio oscillante convoglia il gruppo acetilico verso il sito attivo della diidrolipoil transacetilasi per il definitivo trasferimento al coenzima A. Il braccio di lipoil-lisina può quindi spostarsi sequenzialmente dal sito attivo della piruvato deidrogenasi a quello della diidrolipoil transacetilasi e infine a quello della diidrolipoil deidrogenasi.[2]
Proprietà della tiamina pirofosfato
Nella molecola della tiamina pirofosfato è possibile individuare tre parti distinte, da cui dipendono le sue proprietà chimiche ed enzimologiche: l’anello tiazolico, l’anello 4′-amminopirimidinico e la catena laterale difosfato.[37]
La catena laterale difosfato ancora il cofattore all’enzima attraverso la formazione di legami elettrostatici tra le cariche negative portate dai suoi gruppi fosforici e quelle positive degli ioni Ca2+ e Mg2+ che sono a loro volta legati a sequenze altamente conservate, Gly-Asp-Gly (GDG) per il Ca2+ e Gly-Asp-Gly-X26-Asn-Asn (GDG-X26-NN) per lo Mg2+.[38]
L’anello tiazolico ha un ruolo centrale nella catalisi grazie alla capacità di formare un carbanione, ossia un centro nucleofilo sul C-2.[39]
Nota: come detto in precedenza, una volta legatasi all’enzima, la TPP si dispone nel sito attivo in modo che l’anello tiazolico sia posizionato vicino all’ingresso del canale che porta al sito attivo.[5]
L’anello amminopirimidinico ha una duplice funzione:
- ancora il coenzima tenendolo in posizione;
- ha uno specifico ruolo catalitico, partecipando a una catalisi acido–base. Questo è evidenziato da studi condotti utilizzando analoghi della tiamina pirofosfato in cui, a turno, erano stati sostituiti i tre atomi di azoto dell’anello. Gli studi hanno dimostrato che l’atomo N-1′ e il gruppo amminico legato a N-4′ sono necessari per l’attività del coenzima, mentre l’atomo N-3′ lo è meno.[4]
Come si forma il carbanione dipolare della tiamina pirofosfato
È possibile individuare tre forme tautomeriche in cui si presenta l’anello amminopirimidinico della tiamina pirofosfato legata allo specifico enzima ancora non impegnato nella reazione:
- la forma canonica, ossia la 4′-amminopirimidina;
- la forma protonata su N-1′, ossia lo ione 4′-amminopirimidinio;
- l’1′,4′-imminopirimidina.[40]
Sembra che la 1′,4′-imminopirimidina sia il tautomero che, prima dell’arrivo del substrato nel sito attivo, subisce la deprotonazione.
Il C-2 dell’anello tiazolico è assai più acido rispetto alla maggior parte degli altri gruppi =C–H presenti in altre molecole. La sua inusuale acidità, ossia il fatto che il protone legato al C-2 sia facilmente dissociabile, è dovuta alla presenza dell’atomo di azoto quaternario con carica positiva dell’anello tiazolico che è in grado di stabilizzare elettrostaticamente il carbanione risultante.[41]
Nel processo di deprotonazione sembra avere un ruolo essenziale il gruppo amminico dell’anello amminopirimidinico. Il gruppo funge infatti da base ed è posizionato opportunamente per accettare il protone. Tuttavia, nella forma canonica, la 4′-amminopirimidina, uno dei suoi protoni collide stericamente con il protone legato al C-2. In aggiunta, anche il suo pKa è troppo basso per operare la deprotonazione in modo efficiente.[37]
È stato quindi proposto un meccanismo in cui la catena laterale di un residuo conservato di glutammato, ad esempio βGlu59 in B. stearothermophilus o Glu51 nella piruvato decarbossilasi di Saccharomyces uvarum (lievito di birra), donando un protone all’amminopirimidina la converte nella sua forma imminotautomerica, la 1′,4′-imminopirimidina, che, accettando il protone dal C-2, torna alla forma canonica 4′-amminopirimidinica e permette la formazione del carbanione.[2]
Nota: la formazione del carbanione sul C-2 è quindi conseguenza di un trasferimento protonico intramolecolare.
Deprotonazione della tiamina pirofosfato
La perdita del protone dal C-2 dell’anello tiazolico porta, da una situazione con carica positiva sull’anello, alla formazione di uno ione dipolare o zwitterione. Questo cambio nello stato di carica innesca una modificazione conformazionale in una delle due anse peptidiche conservate presenti all’ingresso del canale che immette al sito attivo, nello specifico la più interna, modificazione che, a sua volta, porta alla chiusura del canale rispetto all’ambiente acquoso circostante. In questa conformazione chiusa, il carbanione tiazolico è protetto dall’attacco di agenti elettrofili.[42]
Riassumendo, la deprotonazione della tiamina pirofosfato determina la chiusura del sito attivo e la protezione del carbanione dipolare neoformato; in altri termini, gli enzimi TPP-dipendenti sarebbero attivi solo nella conformazione chiusa.
Se invece si considera l’altro sito attivo (poiché questi enzimi hanno in genere due siti attivi per dimero), la tiamina pirofosfato non è in forma di ilide, il canale è aperto, e il sito stesso risulta inattivo.[5]
Reazione della diidrolipoil transacetilasi
La diidrolipoil transacetilasi catalizza la terza reazione della sequenza delle cinque catalizzate dai componenti del complesso della piruvato deidrogenasi. La reazione comporta il trasferimento del gruppo acetilico dall’acetil-diidrolipoammide al coenzima A, a dare acetil-CoA e la forma completamente ridotta della lipoammide, la diidrolipoammide, il ditiolo.
Va notato che il gruppo acetilico, inizialmente legato tramite legame tioestere a uno dei gruppi –SH della lipoammide, è di seguito legato al gruppo –SH del coenzima A, di nuovo attraverso legame tioestere, da cui il termine di transtioesterificazione.[4]
Meccanismo di reazione
Nel corso della reazione, il gruppo sulfidrilico del coenzima A porta un attacco nucleofilo al carbonio carbonilico del gruppo acetilico dell’acetil-diidrolipoammide–diidrolipoil transacetilasi a formare un intermedio tetraedrico transiente che si decompone a dare diidrolipoammide–diidrolipoil transacetilasi e acetil-CoA.
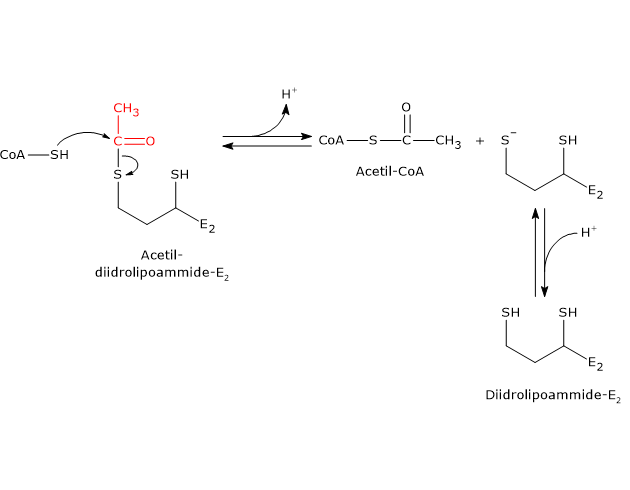
Come già detto, nel meccanismo di reazione gioca un ruolo centrale la mobilità della lipoammide.[43][44]
Reazione della diidrolipoil deidrogenasi
La diidrolipoil deidrogenasi catalizza la quarta e la quinta reazione della sequenza delle cinque catalizzate dai componenti del complesso della piruvato deidrogenasi. L’enzima catalizza il trasferimento degli elettroni necessari a rigenerare il ponte disolfuro della lipoammide della diidrolipoil transacetilasi, ossia, per rigenerare la forma ossidata del gruppo prostetico e completare così il ciclo catalitico della transacetilasi.[5]
La reazione segue un meccanismo catalitico a ping-pong, procedendo attraverso due semireazioni consecutive nelle quali i due substrati, la diidrolipoammide e il NAD+, reagiscono uno in assenza dell’altro. Durante la prima semireazione il rilascio del primo prodotto e la formazione di un complesso enzima–intermedio si verificano prima che il secondo substrato si leghi, mentre l’enzima va incontro a un cambio strutturale. Nella seconda semireazione, si verificano il rilascio del secondo prodotto e il ritorno dell’enzima allo stato iniziale, di nuovo attraverso una modificazione strutturale.[2]
Considerando la cinetica del meccanismo a ping-pong:
- nella prima semireazione si verifica l’ossidazione della diidrolipoammide a lipoammide;
- nella seconda semireazione si verifica la riduzione del NAD+ a NADH.[4][45]
Meccanismo catalitico: prima semireazione
Di seguito viene descritto il meccanismo di reazione della diidrolipoil deidrogenasi di P. putida.
Nella prima semireazione l’enzima in forma ossidata (E), ossia con il ponte disolfuro Cys43–Cys48, lega la diidrolipoammide (LH2) formando il complesso enzima−diidrolipoammide (E–LH2). A questo punto, uno zolfo della diidrolipoammide porta un attacco nucleofilo allo zolfo della Cys43, con formazione di un ponte disolfuro lipoammide–Cys43, (E–S–S–L), mentre lo zolfo della Cys48 viene rilasciato in forma di ione tiolato (S48–).[34]
Il protone residuo sul secondo gruppo tiolico della lipoammide è quindi estratto ad opera dell’istidina (His) 451, che agisce da catalizzatore acido-base generale, determinando la formazione di un secondo ione tiolato sulla lipoammide (E–S–S–L–S–). Questo tiolato, attraverso un attacco nucleofilo, va a sostituire lo zolfo della Cys43, S43, aiutato in questo attacco dalla catalisi acida generale ad opera della His451, che dona un protone a S43. L’azione catalitica della His451 è essenziale, come dimostrato da esperimenti di mutagenesi sito-specifica: la sua sostituzione con glutammina riduce l’attività enzimatica a circa lo 0,4% rispetto al wild-type.[18]
L’anione tiolato S48− entra quindi in contatto, mediante interazioni non covalenti, con l’anello flavinico in prossimità della posizione 4a. Una coppia di elettroni di S48−, il donatore di elettroni, è parzialmente trasferita all’anello flavinico ossidato, l’accettore di elettroni. La struttura derivante è chiamata complesso a trasferimento di carica.
Nel frattempo, la catena fenolica della Tyr181 continua a schermare l’anello flavinico, proteggendolo dall’ossidazione da parte di O2.[2]
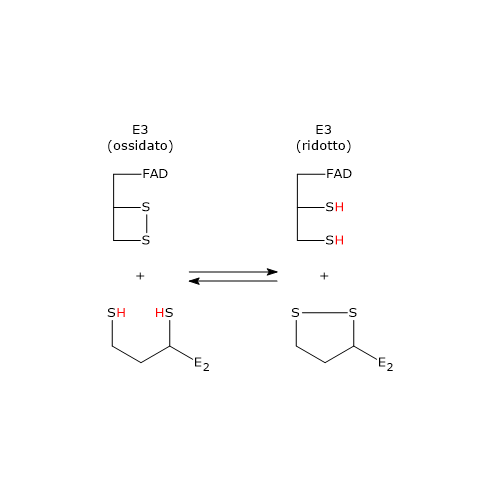
Quello che si verifica è quindi una reazione di interscambio di ponti disolfuro. I prodotti sono la forma ossidata della lipoammide, rilasciata come primo prodotto, e la forma ridotta della diidrolipoil deidrogenasi.
Meccanismo catalitico: seconda semireazione
La seconda semireazione comporta la riduzione del NAD+ a NADH + H+ mediante il trasferimento di elettroni dal disolfuro reattivo dell’enzima via FAD.
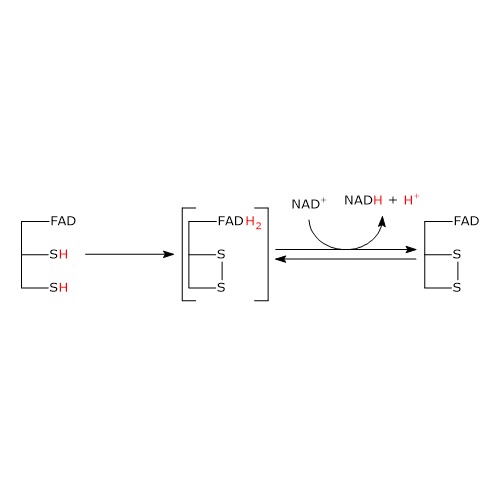
Il tutto ha inizio con l’ingresso nel sito attivo del NAD+ e il suo legame a formare il complesso E−H2−NAD+. Si noti che l’ingresso del coenzima determina lo spostamento a lato della catena laterale fenolica della Tyr181 ad opera dell’anello nicotinammidico.
A seguito del collasso del complesso a trasferimento di carica, si forma un legame covalente tra il C-4a dell’anello flavinico e S48, accompagnato dall’estrazione di un protone da parte dell’N-5 flavinico, con formazione del corrispondente anione tiolato S43–.
S43– porta quindi un attacco nucleofilo a S48, che determina la formazione del ponte disolfuro redox-attivo tra la Cys43 e la Cys48, cui segue la rottura del legame covalente tra S48 e il C-4a dell’anello flavinico a dare l’anione FADH–, con carica negativa su N-1.
Si noti che la diidrolipoil deidrogenasi è nuovamente nella forma ossidata (E).[34]
Il FADH– ha un’esistenza transitoria in quanto si verifica il trasferimento praticamente immediato del protone legato a N-5, in forma di ione idruro, al C-4 dell’anello nicotinammidico, che è giustapposto all’N-5 flavinico. Questo porta alla formazione di FAD e del secondo prodotto della reazione, il NADH, che lascia l’enzima.[1]
Implicazioni funzionali e confronto con la glutatione reduttasi
In definitiva, ciò che accade è che gli elettroni rimossi dal gruppo idrossietilico, derivato dal piruvato, passano via FAD al NAD+. In questo modo si è completato il ciclo catalitico della diidrolipoil deidrogenasi, essendo l’enzima e il suo coenzima nella loro forma ossidata. A questo punto anche il ciclo catalitico dell’intero complesso della piruvato deidrogenasi si è completato, e il complesso è pronto per un altro ciclo di reazioni.[1]
Nota: a differenza dell’anello tiazolico della tiamina pirofosfato, il FAD non ha la funzione di trappola o pozzo per gli elettroni quanto piuttosto di condotta per gli elettroni tra il disolfuro redox-attivo nella sua forma ridotta e il NAD+.
Il meccanismo catalitico della diidrolipoil deidrogenasi è stato in gran parte determinato in analogia con quello della glutatione reduttasi (EC 1.8.1.7), al 33% identica ma strutturalmente molto meglio caratterizzata.[46] Va tuttavia notato che, sebbene i due enzimi catalizzino reazioni simili, queste normalmente procedono in direzioni opposte:
- la diidrolipoil deidrogenasi utilizza il NAD+ per ossidare due gruppi −SH a dare un disolfuro (−S−S−);
- la glutatione reduttasi utilizza il NADPH per ridurre un −S−S− a due gruppi tiolici.
Nonostante ciò, i siti attivi dei due enzimi sono strettamente sovrapponibili.[2]
Regolazione dell’attività del complesso della piruvato deidrogenasi
Nei mammiferi, la regolazione dell’attività del complesso della piruvato deidrogenasi è essenziale sia nello stato di digiuno che in quello alimentato. Infatti, il complesso multienzimatico svolge un ruolo centrale nel metabolismo in quanto, catalizzando la decarbossilazione ossidativa irreversibile del piruvato, rappresenta la porta di ingresso dello scheletro carbonioso dei carboidrati e di circa il 50% dello scheletro carbonioso degli amminoacidi glucogenici, che nell’insieme costituiscono circa il 60% dell’apporto calorico giornaliero, verso:
- il ciclo dell’acido citrico, e quindi alla completa ossidazione a CO2;
- la sintesi dei lipidi, nello stato alimentato, e dell’acetilcolina.[47]
L’importanza della regolazione della conversione del piruvato in acetil-CoA è sottolineata anche dal fatto che i mammiferi, sebbene siano in grado di produrre glucosio dal piruvato, non sono in grado di farlo dall’acetil-CoA. Ciò è conseguenza sia della irreversibilità della reazione catalizzata dalla piruvato deidrogenasi che dell’assenza di vie metaboliche alternative in grado di farlo. L’inibizione dell’attività del complesso permetterà quindi di risparmiare glucosio e gli amminoacidi che possono essere convertiti in piruvato, come l’alanina, quando sono disponibili altri carburanti, ad esempio l’acetil-CoA derivante dall’ossidazione degli acidi grassi.[48]
Per questi motivi l’attività del complesso è finemente regolata attraverso:
- inibizione da feedback, anche nota come inibizione da prodotto finale;
- nucleotidi;
- modificazioni covalenti, ossia fosforilazioni e defosforilazioni di specifiche proteine bersaglio.[5]
Regolazione attraverso inibizione da prodotto finale e stato energetico della cellula
L’attività della forma defosforilata del complesso della piruvato deidrogenasi è regolata attraverso inibizione da feedback o inibizione da prodotto finale.
Acetil-CoA e NADH inibiscono allostericamente gli enzimi che ne catalizzano la sintesi, rispettivamente la diidrolipoil transacetilasi e la diidrolipoil deidrogenasi.[49]
Inoltre, il CoA e l’acetil-CoA, così come il NAD+ e il NADH, competono per i siti di legame sui rispettivi enzimi, quindi E2 ed E3, i quali catalizzano reazioni reversibili. Questo significa che, in presenza di elevati valori dei rapporti [NADH]/[NAD+] e [Acetil-CoA]/[CoA], le reazioni di transacetilazione e deidrogenazione vanno nella direzione opposta rispetto a quella della formazione dell’acetil-CoA. Di conseguenza, la diidrolipoil transacetilasi non può accettare il gruppo idrossietilico dalla TPP in quanto è mantenuta nella forma acetilata. Questo fa sì che la tiamina pirofosfato rimanga legata alla piruvato deidrogenasi nella sua forma idrossietilica, il che, a sua volta, riduce la velocità di decarbossilazione del piruvato. Quindi, elevati valori dei rapporti [NADH]/[NAD+] e [Acetil-CoA]/[CoA] influenzano indirettamente l’attività della piruvato deidrogenasi.[1]
Collegamenti con l’ossidazione degli acidi grassi
Acetil-CoA e NADH sono prodotti anche durante l’ossidazione degli acidi grassi che, al pari delle reazioni catalizzate dal complesso della piruvato deidrogenasi, si verifica nel mitocondrio. Ciò significa che la cellula, attraverso la regolazione dell’attività del complesso multienzimatico, è in grado di preservare le riserve di carboidrati quando sono disponibili acidi grassi per la produzione di energia.
Questa relazione reciproca tra l’ossidazione del glucosio e quella degli acidi grassi, caratterizzata dall’inibizione dell’utilizzo del glucosio quando la disponibilità di acidi grassi è elevata, è nota come ciclo di Randle (o ciclo glucosio-acidi grassi). Questo per esempio è quanto accade nel digiuno, quando il fegato, il muscolo scheletrico e molti altri organi e tessuti si affidano principalmente all’ossidazione degli acidi grassi per la produzione di energia. Di contro, l’attività del complesso è aumentata nello stato alimentato, quando molti differenti tipi di cellule e tessuti utilizzano in prevalenza il glucosio come fonte di energia.
Più in generale, quando la produzione di NADH e/o acetil-CoA supera la capacità della cellula di utilizzarli per la produzione di ATP, l’attività del complesso della piruvato deidrogenasi è inibita. Analogo discorso vale nella condizione in cui non ci sia necessità di produrre ulteriore ATP. Infatti, l’attività catalitica del complesso multienzimatico è sensibile anche allo stato energetico della cellula. Attraverso meccanismi allosterici, elevati livelli di ATP inibiscono l’attività della componente piruvato deidrogenasi del complesso multienzimatico, mentre elevati livelli di ADP, che segnalano che la cellula potrebbe entrare in una fase di carenza di energia, lo attivano, indirizzando quindi lo scheletro carbonioso dei carboidrati e di alcuni amminoacidi verso la produzione di energia.
Nota: nel muscolo scheletrico l’attività del complesso della piruvato deidrogenasi aumenta con l’aumento dell’attività aerobica. Questo si traduce in una maggiore dipendenza del muscolo dal glucosio come fonte di energia.[4]
Regolazione attraverso fosforilazione/defosforilazione
A differenza di quanto accade nei procarioti, nei mammiferi l’attività del complesso della piruvato deidrogenasi è regolata anche attraverso modificazioni covalenti. Nello specifico, si tratta di fosforilazioni e defosforilazioni di tre specifici residui di serina presenti sulla subunità α della piruvato deidrogenasi, l’enzima che catalizza il primo passaggio, irreversibile, dell’intera sequenza di reazioni.
Nota: poiché la piruvato deidrogenasi dei mammiferi è un eterotetramero, ci sono sei potenziali siti di fosforilazione.[1]
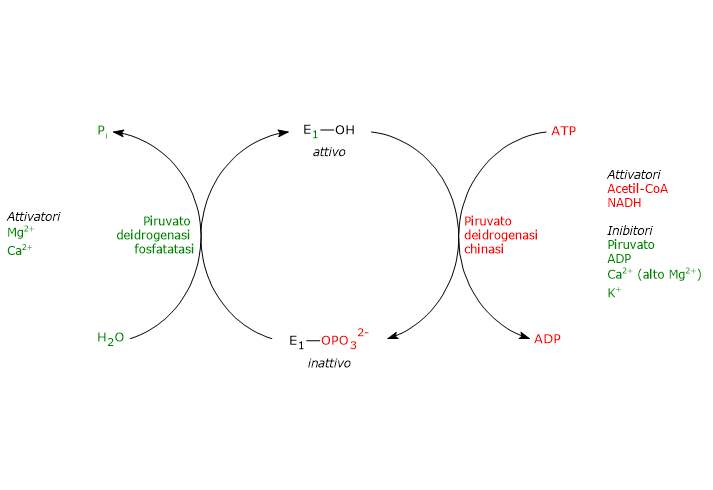
La fosforilazione, che inattiva la piruvato deidrogenasi e quindi blocca l’intera sequenza di reazioni, è catalizzata dalla piruvato deidrogenasi chinasi, enzima Mg2+-dipendente. Due delle suddette serine si trovano su una delle due anse presenti all’ingresso del canale per il substrato che porta al rispettivo sito attivo, quella più vicina all’estremità C-terminale. La fosforilazione di uno solo di questi residui inattiva la piruvato deidrogenasi, dimostrando così l’accoppiamento fuori fase tra i due siti attivi.[50][51]
Nello stato defosforilato invece il complesso risulta attivo. La defosforilazione è catalizzata da una specifica protein fosfatasi, la piruvato deidrogenasi fosfatasi, enzima attivato dal Ca2+.[52]
Le attività della piruvato deidrogenasi chinasi e della piruvato deidrogenasi fosfatasi sono a loro volta soggette a regolazione allosterica da parte di numero effettori.[53]
Regolazione della piruvato deidrogenasi chinasi
L’attività della piruvato deidrogenasi chinasi dipende dai rapporti [NADH]/[NAD+], [acetil-CoA]/[CoA], e [ATP]/[ADP], nonché dalla concentrazione del piruvato nella matrice mitocondriale.[54]
- Elevati valori dei rapporti [NADH]/[NAD+] e [acetil-CoA]/[CoA], come durante l’ossidazione degli acidi grassi e dei corpi chetonici, attivano la chinasi. La piruvato deidrogenasi viene quindi fosforilata, e il complesso multienzimatico risulta inibito. Questo consente a tessuti come il muscolo cardiaco di risparmiare glucosio quando si stanno utilizzando acidi grassi e/o corpi chetonici per la produzione di energia, poiché la sintesi di acetil-CoA dal piruvato, e quindi dai carboidrati (e da alcuni amminoacidi) viene bloccata. Quando invece le concentrazioni di NAD+ e coenzima A sono elevate, la chinasi è inibita e il complesso risulta attivo. Quindi, acetil-CoA e NADH, due dei tre prodotti finali delle reazioni catalizzate dal complesso della piruvato deidrogenasi, regolano la propria sintesi esercitando un controllo allosterico diretto e indiretto sull’attività del complesso.
- Elevati valori del rapporto [ATP]/[ADP] attivano la chinasi e quindi inibiscono il complesso multienzimatico. A differenza di molte altre chinasi, come quelle coinvolte nel controllo del metabolismo del glicogeno, la piruvato deidrogenasi chinasi non è regolata dai livelli di cAMP, ma da molecole che segnalano variazioni nel livello della carica energetica cellulare e nella disponibilità di intermedi biosintetici, ossia rispettivamente ATP e NADH e acetil-CoA.
- Il piruvato è un effettore allosterico negativo della piruvato deidrogenasi chinasi. Quando i suoi livelli sono elevati, il suo legame alla chinasi la inattiva, la piruvato deidrogenasi non viene fosforilata, e il complesso della piruvato deidrogenasi rimane attivo.
- La piruvato deidrogenasi chinasi è attivata anche a seguito dell’interazione con la diidrolipoil transacetilasi nella sua forma acetilata, ossia quando è presente l’acetil-diidrolipoammide.
Altri attivatori della piruvato deidrogenasi chinasi sono gli ioni potassio e magnesio.[2][53]
Regolazione della piruvato deidrogenasi fosfatasi
L’attività della piruvato deidrogenasi fosfatasi dipende dal valore dei rapporti [NADH]/[NAD+] e [acetil-CoA]/[CoA], come anche dalla concentrazione degli ioni Ca2+ nella matrice mitocondriale.
- Bassi valori dei rapporti [NADH]/[NAD+] e [acetil-CoA]/[CoA] attivano la fosfatasi, la piruvato deidrogenasi viene defosforilata, e il complesso multienzimatico risulta attivo. Al contrario, in presenza di elevati valori dei suddetti rapporti l’attività della fosfatasi si riduce, quella della chinasi aumenta, e il complesso multienzimatico viene inibito.
- Lo ione calcio attiva la piruvato deidrogenasi fosfatasi.
Il Ca2+ è un importante secondo messaggero che segnala la necessità da parte della cellula di ulteriore energia. Quindi, quando è presente in elevate concentrazioni, come nelle cellule muscolari cardiache a seguito della stimolazione da parte dell’adrenalina, o nelle cellule muscolari scheletriche nel corso della contrazione muscolare, la fosfatasi è attiva, il complesso è defosforilato e quindi attivato. - Anche l’insulina interviene nel controllo dell’attività catalitica del complesso della piruvato deidrogenasi attraverso l’attivazione della piruvato deidrogenasi fosfatasi. L’ormone, in risposta all’aumento della glicemia, stimola sia la sintesi del glicogeno che dell’acetil-CoA, precursore nella sintesi dei lipidi.[2][53]
Anche il digiuno e la successiva rialimentazione influiscono sull’attività del complesso multienzimatico.
In tessuti come il muscolo scheletrico, il muscolo cardiaco o il rene, il digiuno riduce in modo significativo l’attività del complesso, mentre la rialimentazione inverte la situazione. Nel cervello invece non si osservano queste variazioni poiché l’attività del complesso della piruvato deidrogenasi è essenziale per la produzione di ATP.[1][55]
Bibliografia
- ^ a b c d e f g h i j Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
- ^ a b c d e f g h i j k l m n o Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
- ^ a b c Patel M.S., Nemeria N.S., Furey W., Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem 2014;289(24):16615-16623. doi:10.1074/jbc.R114.563148
- ^ a b c d e f g h i j k Berg J.M., Tymoczko J.L., Gatto G.J. Jr., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
- ^ a b c d e f g h i j k Garrett R.H., Grisham C.M. Biochemistry. 7th Edition. Cengage Learning, 2023.
- ^ a b c d Bothe S.N., Zdanowicz R. Structural diversity of pyruvate dehydrogenase complexes. FEBS Lett 2025. doi:10.1002/1873-3468.70140
- ^ Czumaj A., Szrok-Jurga S., Hebanowska A., Turyn J., Swierczynski J., Sledzinski T., Stelmanska E. The pathophysiological role of CoA. Int J Mol Sci 2020;21(23):9057. doi:10.3390/ijms21239057
- ^ Solomons T.W.G., Fryhle C.B., Snyder S.A. Solomons’ organic chemistry. 12th Edition. John Wiley & Sons Incorporated, 2017.
- ^ Solmonson A., DeBerardinis R.J. Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem 2018;293(20):7522-7530. doi:10.1074/jbc.TM117.000259
- ^ a b Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 4th Edition. St. Louis: Elsevier, 2018.
- ^ Lu Z., Imlay J.A. When anaerobes encounter oxygen: mechanisms of oxygen toxicity, tolerance and defence. Nat Rev Microbiol 2021;19(12):774-785. doi:10.1038/s41579-021-00583-y
- ^ Chaudhry R., Varacallo M.A. Biochemistry, Glycolysis. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482303/
- ^ Zangari J., Petrelli F., Maillot B., Martinou J.C. The multifaceted pyruvate metabolism: role of the mitochondrial pyruvate carrier. Biomolecules 2020;10(7):1068. doi:10.3390/biom10071068
- ^ Reed L.J., Pettit F.H., Eley M.H., Hamilton L., Collins J.H., Oliver R.M. Reconstitution of the Escherichia coli pyruvate dehydrogenase complex. Proc Natl Acad Sci USA. 1975;72(8):3068-72. doi:10.1073/pnas.72.8.3068
- ^ Stoops J.K., Cheng R.H., Yazdi M.A., et al. On the unique structural organization of the Saccharomyces cerevisiae pyruvate dehydrogenase complex. J Biol Chem 1997;272(9):5757-64. doi:10.1074/jbc.272.9.5757
- ^ a b Zhou Z.H., McCarthy D.B., O’Connor C.M., Reed L.J., Stoops J.K. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA 2001;98(26):14802-7. doi:10.1073/pnas.011597698
- ^ Forsberg B.O., Aibara S., Howard R.J., Mortezaei N., Lindahl E. Arrangement and symmetry of the fungal E3BP-containing core of the pyruvate dehydrogenase complex. Nat Commun 2020;11(1):4667. doi:10.1038/s41467-020-18401-z
- ^ a b Ciszak E.M., Makal A., Hong Y.S., Vettaikkorumakankauv A.K., Korotchkina L.G., Patel M.S. How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J Biol Chem 2006;281(1):648-55. doi:10.1074/jbc.M507850200
- ^ a b Wang C., Ma C., Xu Y., et al. Dynamics of the mammalian pyruvate dehydrogenase complex revealed by in-situ structural analysis. Nat Commun 2025;16(1):917. doi:10.1038/s41467-025-56171-8
- ^ Milne J.L., Wu X., Borgnia M.J., Lengyel J.S., Brooks B.R., Shi D., Perham R.N., Subramaniam S. Molecular structure of a 9-MDa icosahedral pyruvate dehydrogenase subcomplex containing the E2 and E3 enzymes using cryoelectron microscopy. J Biol Chem 2006;281(7):4364-70. doi:10.1074/jbc.M504363200
- ^ Kale S., Arjunan P., Furey W., Jordan F. A dynamic loop at the active center of the Escherichia coli pyruvate dehydrogenase complex E1 component modulates substrate utilization and chemical communication with the E2 component. J Biol Chem 2007;282(38):28106-16. doi:10.1074/jbc.M704326200
- ^ a b c Frank R.A.W., Titman C.M., Pratap J.V., Luisi B.F., Perham R.N. A molecular switch and proton wire synchronize the active sites in thiamine enzymes. Science 2004;306(5697):872-876. doi:10.1126/science.1101030
- ^ a b c Nemeria N.S., Arjunan P., Chandrasekhar K., Mossad M., Tittmann K., Furey W., Jordan F. Communication between thiamin cofactors in the Escherichia coli pyruvate dehydrogenase complex E1 component active centers: evidence for a “direct pathway” between the 4′-aminopyrimidine N1′ atoms. J Biol Chem 2010;285(15):11197-209. doi:10.1074/jbc.M109.069179
- ^ Arjunan P., Nemeria N., Brunskill A., Chandrasekhar K., Sax M., Yan Y., Jordan F., Guest J.R., Furey W. Structure of the pyruvate dehydrogenase multienzyme complex E1 component from Escherichia coli at 1.85 A resolution. Biochemistry 2002;41(16):5213-21. doi:10.1021/bi0118557
- ^ a b Perham R.N. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem 2000;69:961-1004. doi:10.1146/annurev.biochem.69.1.961
- ^ a b Wang J., Nemeria N.S., Chandrasekhar K., et al. Structure and function of the catalytic domain of the dihydrolipoyl acetyltransferase component in Escherichia coli pyruvate dehydrogenase complex. J Biol Chem 2014;289(22):15215-30. doi:10.1074/jbc.M113.544080
- ^ Allen A.G., Perham R.N. Two lipoyl domains in the dihydrolipoamide acetyltransferase chain of the pyruvate dehydrogenase multienzyme complex of Streptococcus faecalis. FEBS Lett 1991;287(1-2):206-10. doi:10.1016/0014-5793(91)80052-5
- ^ Hanemaaijer R., de Kok A., Jolles J., Veeger C. The domain structure of the dihydrolipoyl transacetylase component of the pyruvate dehydrogenase complex from Azotobacter vinelandii. Eur J Biochem 1987;169(2):245-52. doi:10.1111/j.1432-1033.1987.tb13604.x
- ^ Hale G., Perham R.N. Amino acid sequence around lipoic acid residues in the pyruvate dehydrogenase multienzyme complex of Escherichia coli. Biochem J 1980;187(3):905-8. doi:10.1042/bj1870905
- ^ Hipps D.S., Packman L.C., Allen M.D., Fuller C., Sakaguchi K., Appella E., Perham R.N. The peripheral subunit-binding domain of the dihydrolipoyl acetyltransferase component of the pyruvate dehydrogenase complex of Bacillus stearothermophilus: preparation and characterization of its binding to the dihydrolipoyl dehydrogenase component. Biochem J 1994;297(Pt 1):137-43. doi:10.1042/bj2970137
- ^ Schulze E., Westphal A.H., Obmolova G., Mattevi A., Hol W.G., de Kok A. The catalytic domain of the dihydrolipoyl transacetylase component of the pyruvate dehydrogenase complex from Azotobacter vinelandii and Escherichia coli. Expression, purification, properties and preliminary X-ray analysis. Eur J Biochem 1991;201(3):561-8. doi:10.1111/j.1432-1033.1991.tb16315.x
- ^ Duarte I.F., Caio J., Moedas M.F., Rodrigues L.A., Leandro A.P., Rivera I.A., Silva M.F.B. Dihydrolipoamide dehydrogenase, pyruvate oxidation, and acetylation-dependent mechanisms intersecting drug iatrogenesis. Cell Mol Life Sci 2021;78(23):7451-7468. doi:10.1007/s00018-021-03996-3
- ^ Brautigam C.A., Chuang J.L., Tomchick D.R., Machius M., Chuang D.T. Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations. J Mol Biol 2005;350(3):543-52. doi:10.1016/j.jmb.2005.05.014
- ^ a b c Mattevi A., Obmolova G., Sokatch J.R., Betzel C., Hol W.G. The refined crystal structure of Pseudomonas putida lipoamide dehydrogenase complexed with NAD+ at 2.45 A resolution. Proteins 1992;13(4):336-51. doi:10.1002/prot.340130406
- ^ Kern D., Kern G., Neef H., Tittmann K., Killenberg-Jabs M., Wikner C., Schneider G., Hübner G. How thiamine diphosphate is activated in enzymes. Science 1997;275(5296):67-70. doi:10.1126/science.275.5296.67
- ^ a b Jordan F. Current mechanistic understanding of thiamin diphosphate-dependent enzymatic reactions. Nat Prod Rep 2003;20(2):184-201. doi:10.1039/b111348h
- ^ a b Nemeria N.S., Chakraborty S., Balakrishnan A., Jordan F. Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at individual steps. FEBS J 2009;276(9):2432-46. doi:10.1111/j.1742-4658.2009.06964.x
- ^ Hawkins C.F., Borges A., Perham R.N. A common structural motif in thiamin pyrophosphate-binding enzymes. FEBS Lett 1989;255(1):77-82. doi:10.1016/0014-5793(89)81064-6
- ^ Breslow R. Rapid deuterium exchange in thiazolium salts. J Am Chem Soc 1957;79:1762-1763. doi:10.1021/ja01564a064
- ^ Baykal A.T., Kakalis L., Jordan F. Electronic and nuclear magnetic resonance spectroscopic features of the 1′,4′-iminopyrimidine tautomeric form of thiamin diphosphate, a novel intermediate on enzymes requiring this coenzyme. Biochemistry 2006;45(24):7522-8. doi:10.1021/bi060395k
- ^ Meyer D., Neumann P., Koers E., Sjuts H., Lüdtke S., Sheldrick G.M., Ficner R., Tittmann K. Unexpected tautomeric equilibria of the carbanion-enamine intermediate in pyruvate oxidase highlight unrecognized chemical versatility of thiamin. Proc Natl Acad Sci USA 2012;109(27):10867-72. doi:10.1073/pnas.1201280109
- ^ Shaanan B., Chipman D.M. Reaction mechanisms of thiamin diphosphate enzymes: new insights into the role of a conserved glutamate residue. FEBS J 2009 May;276(9):2447-53. doi:10.1111/j.1742-4658.2009.06965.x
- ^ Yang Y.S., Frey P.A. Dihydrolipoyl transacetylase of Escherichia coli. Formation of 8-S-acetyldihydrolipoamide. Biochemistry 1986;25(25):8173-8. doi:10.1021/bi00373a008
- ^ Škerlová J., Berndtsson J., Nolte H., Ott M., Stenmark P. Structure of the native pyruvate dehydrogenase complex reveals the mechanism of substrate insertion. Nat Commun 2021;12(1):5277. doi:10.1038/s41467-021-25570-y
- ^ Massey V., Gibson Q.H., Veeger C. Intermediates in the catalytic action of lipoyl dehydrogenase (diaphorase). Biochem J 1960;77(2):341-51. doi:10.1042/bj0770341
- ^ Rice D.W., Schulz G.E., Guest J.R. Structural relationship between glutathione reductase and lipoamide dehydrogenase. J Mol Biol 1984;174(3):483-96. doi:10.1016/0022-2836(84)90332-2
- ^ Denton R.M., Randle P.J., Bridges B.J., et al. Regulation of mammalian pyruvate dehydrogenase. Mol Cell Biochem 1975;9(1):27-53. doi:10.1007/BF01731731
- ^ Gray L.R., Tompkins S.C., Taylor E.R. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-2604. doi:10.1007/s00018-013-1539-2
- ^ Harris R.A., Bowker-Kinley M.M., Huang B., Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul 2002;42:249-59. doi:10.1016/s0065-2571(01)00061-9
- ^ Kolobova E., Tuganova A., Boulatnikov I., Popov K.M. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J 2001;358(Pt 1):69-77. doi:10.1042/0264-6021:3580069
- ^ Hiromasa Y., Fujisawa T., Aso Y., Roche T.E. Organization of the cores of the mammalian pyruvate dehydrogenase complex formed by E2 and E2 plus the E3-binding protein and their capacities to bind the E1 and E3 components. J Biol Chem 2004;279(8):6921-33. doi:10.1074/jbc.M308172200
- ^ Roche T.E., Baker J.C., Yan X., et al. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol 2001;70:33-75. doi:10.1016/s0079-6603(01)70013-x
- ^ a b c Pettit F.H., Pelley J.W., Reed L.J. Regulation of pyruvate dehydrogenase kinase and phosphatase by acetyl-CoA/CoA and NADH/NAD ratios. Biochem Biophys Res Commun 1975;65(2):575-82. doi:10.1016/s0006-291x(75)80185-9
- ^ Patel M.S., Korotchkina L.G. Regulation of the pyruvate dehydrogenase complex. Biochem Soc T 2006;34(2):217-222. doi:10.1042/bst0340217
- ^ Small L., Brandon A.E., Quek L.E., Krycer J.R., James D.E., Turner N., Cooney G.J. Acute activation of pyruvate dehydrogenase increases glucose oxidation in muscle without changing glucose uptake. Am J Physiol Endocrinol Metab 2018;315(2):E258-E266. doi:10.1152/ajpendo.00386.2017