Carbanions are ions containing a negatively charged carbon atom.
They are formed by the heterolytic cleavage of a covalent bond between a carbon atom and another atom or group.[1]
Having an unshared electron pair, they are powerful nucleophiles, and strong bases, and attack, in order to form a covalent bond, a proton or an electrophilic center, such as a polarized or positively charged center.[2]
Carbanions are extremely reactive. Therefore, they must be stabilized to allow them to attack electrophilic centers.[3] Stabilization may occur by inductive effect, resonance, and may also depend on the hybridization of the carbon atom carrying the negative charge.[1][2]
They are intermediates in many enzyme-catalyzed reactions.
Summary: Key Points
- Reactive Intermediates: carbanions are organic ions containing a negatively charged carbon atom with an unshared electron pair, making them powerful nucleophiles and strong bases.
- Heterolytic cleavage: they are generated via the non-uniform breaking (heterolysis) of a covalent bond, where the carbon atom retains both bonding electrons from a less electronegative group.
- Stabilization mechanisms: as highly transient species, their stability depends on negative inductive effects (-I), resonance delocalization within electron sinks, and an increased s-character of the hybrid orbitals (sp > sp2 > sp3).
- Biochemical roles: They serve as vital intermediates in key enzyme-catalyzed pathways, such as 2-oxoacid oxidative decarboxylations, where their formation is actively mediated and stabilized by the cofactor thiamine pyrophosphate.
Contents
Heterolysis and homolysis
Considering two atoms or groups, indicated as A and B, joined by a covalent bond, there are two ways to break the bond: heterolysis and homolysis.
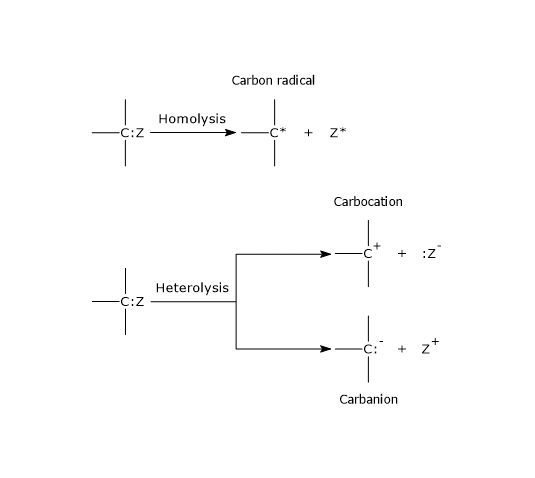
In heterolysis, the breaking of the covalent bond leads to the formation of two charged atoms, namely two ions, a cation and an anion, as both bonding electrons are retained by by the more electronegative of the two atoms.[4]
A:B → :A– + B+, if A is more electronegative than B;
A:B → A+ + :B–, if B is more electronegative than A.
In the heterolysis of a covalent bond involving a carbon atom, if both electrons are retained by the carbon atom, it will have a negative charge, therefore it is an anion, and is defined as a carbanion. On the contrary, if the carbon loses both electrons, it will have a positive charge, therefore it is a cation, and is defined as a carbocation.[5]
In homolysis, the breaking of the covalent bond between A and B leads to the formation of two free radicals, as each atom or group takes one of the two bonding electrons.[6] Free radicals, which are electrically neutral, are also very unstable molecules. Note that homolytic cleavage is less common than heterolytic cleavage.[7]
Stabilization of carbanions
Carbanions are extremely reactive chemical species, and, like carbocations and free radicals, they are almost always transient intermediates in organic reactions. In order to allow their attack to the electrophilic centers, they must be stabilized. Their stabilization depends on the dispersion of the negative charge, which may occur by inductive effect, resonance, and may also depend on the s-character of the hybrid orbitals of the negatively charged carbon atom.
The inductive effect is due to the presence in the molecule of one or more permanent dipoles in one or more bonds, dipoles which in turn arise from the difference in electronegativity between two groups. This difference leads to a non-uniform distribution of the bonding electrons. The inductive effect can be positive, also known as +I effect, feature of atoms or groups that tend to repel electrons, or negative, also known as –I effect, which is characteristic of atoms or groups that tend to attract electrons. The atoms or groups with the +I effect tend to decrease the stability of the carbanions, whereas those with the –I effect, therefore more electronegative, tend to stabilize them.[1]
The stability of carbanions increases when they are bound to an electrophilic structure where the unshared electron pair can delocalize by resonance, therefore a structure that acts as an electron trap or electron sink. Aromatic structures, such as the phenyl group, are particularly effective.[2]
Finally, the stability is also a function of the s-character of hybrid orbitals of the negatively charged carbon atom, increasing as the percentage s character increases. Therefore it will increase going from sp3 hybridization, which has 25% s character, to sp2, with 33% s character, to sp, with 50% s-character.[1]
R-CH2– < R1R2C=CH– < RC≡C–
Carbanions in enzymatic reactions
Examples of enzymatic reactions that proceed with the formation of carbanions are those catalyzed by three multienzyme complexes belonging to the family of 2-oxoacid dehydrogenases or alpha-ketoacid dehydrogenases, which are involved in the oxidative decarboxylation of ketoacids, in particular of α-ketoacids, briefly described below.
- The pyruvate dehydrogenase complex, which catalyzes the oxidative decarboxylation of pyruvate, the conjugate base of pyruvic acid, into acetyl-CoA, thus acting as a bridge between glycolysis and the citric acid cycle;
- The oxoglutarate dehydrogenase or alpha-ketoglutarate dehydrogenase complex, which catalyzes the oxidative decarboxylation of α-ketoglutarate to succinyl-CoA in step 4 of the citric acid cycle;
- The branched-chain α-ketoacid dehydrogenase complex, which catalyzes the oxidative decarboxylation of the branched amino acids valine, leucine and isoleucine into acetyl-CoA and succinyl-CoA. The remaining carbon skeleton can then enter the citric acid cycle.[3]
The three multienzyme complexes have very similar structures and reaction mechanisms, and their E1 subunits, which are thiamine pyrophosphate dependent enzymes, catalyze a reaction in which a carbanion intermediate is formed, whose formation and stabilization by resonance involves thiamine.[8]
Transketolase (EC 2.2.1.1) also catalyzes a reaction that involves the formation of a carbanion intermediate. This enzyme, which catalyzes steps 6 and 8 of the pentose phosphate pathway, requires thiamine pyrophosphate as a cofactor, and has a reaction mechanism similar to that of the E1 subunits of multienzyme complexes seen previously.[7]
Acetyl-CoA carboxylase (EC 6.4.1.2) is another enzyme that catalyzes a reaction that involves the formation of a carbanion intermediate. The enzyme catalyzes the committed step of de novo synthesis of fatty acids, namely, the carboxylation of acetyl-CoA to malonyl-CoA.[9]
References
- ^ a b c d Soderberg T. Organic chemistry with a biological emphasis. Volume I. Chemistry Publications. 2019.
- ^ a b c Solomons T.W.G., Fryhle C.B., Snyder S.A. Solomons’ organic chemistry. 12th Edition. John Wiley & Sons Incorporated, 2017.
- ^ a b Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
- ^ Heterolysis, in IUPAC Compendium of Chemical Terminology, 3rd ed. International Union of Pure and Applied Chemistry; 2006. Online version 3.0.1, 2019. doi:1351/goldbook.H02809.
- ^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
- ^ Homolysis, in IUPAC Compendium of Chemical Terminology, 3rd ed. International Union of Pure and Applied Chemistry; 2006. Online version 3.0.1, 2019. doi:1351/goldbook.H02851.
- ^ a b Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
- ^ Berg J.M., Tymoczko J.L., Gatto J.G., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
- ^ Garrett R.H., Grisham C.M. Biochemistry. 7th Edition. Cengage Learning, 2023.
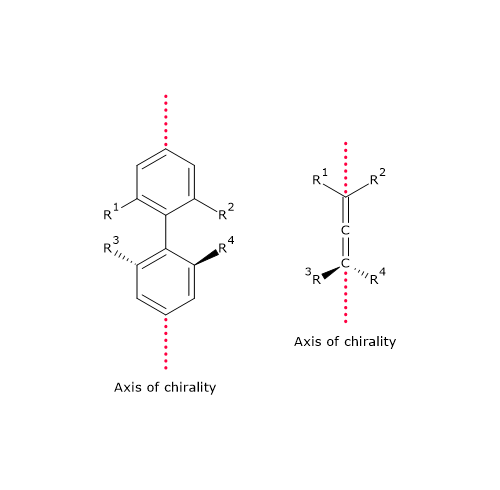 In such cases, chirality results from the presence of an axis of chirality rather than a center. This form of chirality is known as axial chirality.
In such cases, chirality results from the presence of an axis of chirality rather than a center. This form of chirality is known as axial chirality.