Nel 1906, il chimico russo-americano Martin André Rosanoff, allora alla New York University, scelse la gliceraldeide, un monosaccaride, come standard per descrivere la stereochimica di molecole con almeno un centro chirale, come i carboidrati. Questo sistema di nomenclatura divenne noto come convenzione di Fischer-Rosanoff, o convenzione di Rosanoff, o sistema D-L.[1]
In Sintesi: Punti Chiave
- Configurazione relativa: la convenzione di Fischer-Rosanoff (sistema D-L) determina l’orientamento spaziale relativo di carboidrati e amminoacidi prendendo come standard arbitrario la gliceraldeide.
- Indipendenza ottica: non esiste alcuna correlazione diretta tra i prefissi stereochimici D/L (configurazione) e i segni (+)/(–) che indicano la direzione della rotazione ottica.
- Regole di assegnazione: la classificazione si basa sul carbonio asimmetrico più lontano dal gruppo carbonilico nei monosaccaridi, e sul carbonio α asimmetrico negli amminoacidi.
- Limiti strutturali: il sistema definisce unicamente la geometria del carbonio di riferimento; le ambiguità nei sistemi con più centri chirali sono state risolte nel 1956 dalla nomenclatura assoluta RS.
Indice
- Convenzione di Fischer-Rosanoff: origine e applicazioni
- Convenzione di Fischer-Rosanoff: carboidrati
- Convenzione di Fischer-Rosanoff: α-amminoacidi
- Configurazioni relative e assolute
- Limiti della convenzione di Fischer-Rosanoff
- Bibliografia
Convenzione di Fischer-Rosanoff: origine e applicazioni
Poiché all’epoca la configurazione assoluta della gliceraldeide era sconosciuta, Rosanoff assegnò la stereochimica in modo completamente arbitrario.
- Il prefisso D (dal latino dexter, che significa “destra”) fu assegnato alla (+)-gliceraldeide, l’enantiomero destrogiro, assumendo che nella sua proiezioni di Fischer il gruppo ossidrilico (–OH) si trovasse sul lato destro del centro chirale.
- Il prefisso L (dal latino laevus, che significa “sinistra”) fu assegnato alla (–)-gliceraldeide, l’enantiomero levogiro, assumendo che il gruppo ossidrilico fosse sul lato sinistro del centro chirale.[2]
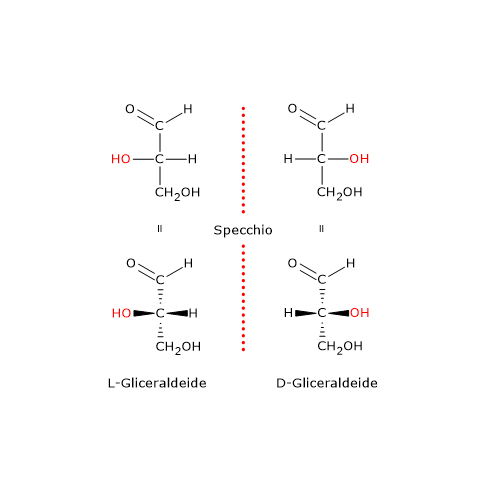
Sebbene lo stesso Emil Fischer avesse rifiutato questo sistema, esso fu ampiamente adottato per determinare le configurazioni relative delle molecole chirali. In che modo? La configurazione attorno a un centro chirale viene messa in relazione con quella della gliceraldeide convertendo chimicamente i gruppi della molecola in esame in quelli del monosaccaride attraverso reazioni che avvengano con ritenzione di configurazione, ossia reazioni che non comportano la rottura di nessuno dei legami del centro chirale. Grazie a ciò la disposizione spaziale dei gruppi attorno al centro chirale nel prodotto è la stessa che si ha nel reagente.[3]
La convenzione di Fischer-Rosanoff consente ai chimici di classificare molecole chirali, come gli amminoacidi e i monosaccaridi, in due categorie: la serie D e la serie L, a seconda che la loro configurazione corrisponda a quella della D- o della L-gliceraldeide.
Nota: non esiste una correlazione diretta tra la configurazione (D o L) e la direzione della rotazione ottica. Il sistema D-L non indica se una molecola è destrogira o levogira; si riferisce soltanto alla configurazione molecolare rispetto alla gliceraldeide.[4]
Convenzione di Fischer-Rosanoff: carboidrati
I monosaccaridi possono essere classificati come aldosi o chetosi. Gli aldosi, e i chetosi con più di tre atomi di carbonio, presentano almeno un centro chirale. Per convenzione, vengono assegnati alla serie D o L a seconda della configurazione del carbonio chirale più distante dal gruppo carbonilico, cioè quello con il più alto stato di ossidazione. Se tale configurazione corrisponde a quella della D- o della L-gliceraldeide, la molecola viene collocata nella rispettiva serie D o L.
Nelle proiezioni di Fischer, la catena carboniosa più lunga è disegnata verticalmente, e gli atomi di carbonio sono numerati in modo che il carbonio del gruppo carbonilico riceva il numero più basso possibile: C-1 negli aldosi e C-2 nei chetosi.[5]
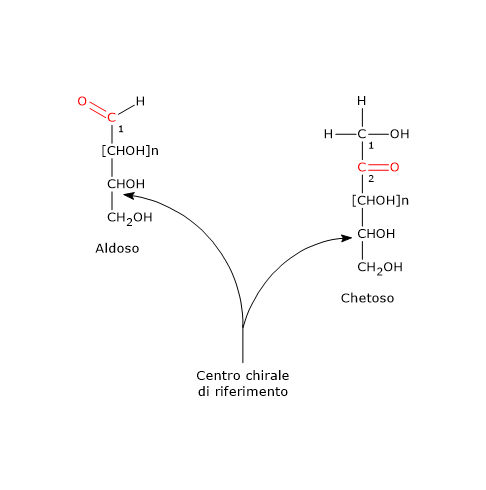
Nota: in Natura, i carboidrati della serie D sono molto più comuni di quelli della serie L.
Se nel nome della molecola deve essere specificato anche il segno della rotazione del piano della luce polarizzata, i prefissi (+) e (–) possono essere utilizzati assieme ai prefissi D e L. Ad esempio, il fruttosio, che è levogiro, può essere scritto come D-(–)-fruttosio, mentre il glucosio, che è destrogiro, come D-(+)-glucosio.
Convenzione di Fischer-Rosanoff: α-amminoacidi
Gli amminoacidi, sulla base della posizione del gruppo amminico (–NH2) rispetto a quella del gruppo carbossilico (–COOH) possono essere classificati come:
- α-amminoacidi, se il gruppo amminico è legato al carbonio α;
- β-amminoacidi, se il gruppo amminico è legato al carbonio β;
- γ-amminoacidi, se il gruppo amminico è legato al carbonio γ;
- δ-amminoacidi, se il gruppo amminico è legato al carbonio δ.
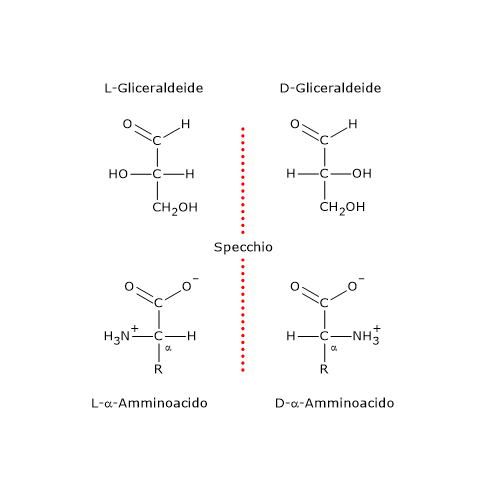
Considerando gli α-amminoacidi, questi apparterranno rispettivamente alla serie D o alla serie L se la configurazione dei gruppi –NH2, –COOH, –R, e dell’atomo di idrogeno legati al carbonio α, il centro chirale, è la stessa di quella dei gruppi –OH, aldeidico (–CHO), idrossimetilico (–CH2OH) e dell’atomo di idrogeno rispettivamente della D-gliceraldeide o della L-gliceraldeide.[4]
Nelle proiezioni di Fischer, gli amminoacidi sono rappresentati con il gruppo carbossilico, il carbonio con il più alto stato di ossidazione, in alto e il gruppo R in basso.
Tra gli α-amminoacidi, quelli proteinogenici, ossia quelli coinvolti nella sintesi proteica, con l’eccezione della glicina, il cui carbonio α non è chirale, presentano tutti la configurazione L, e sono dunque noti come L-α-amminoacidi.
Nota: in natura gli L-α-amminoacidi sono significativamente più abbondanti rispetto agli altri tipi che non partecipano alla sintesi delle proteine.[5]
Configurazioni relative e assolute
Quando Rosanoff assegnò arbitrariamente il prefisso D alla (+)-gliceraldeide e L alla (–)-gliceraldeide, aveva una probabilità del 50% di essere nel giusto.[6]
All’inizio degli anni ’50 lo sviluppo della diffrazione a raggi X rese possibile determinare la configurazione assoluta delle molecole chirali. Nel 1951, il chimico olandese Johannes Martin Bijvoet determinò la configurazione assoluta del (+)-tartrato di rubidio e sodio tetraidrato. Confrontandola con quella della gliceraldeide, dimostrò che l’ipotesi di Rosanoff era corretta.
Di conseguenza, le configurazioni delle molecole chirali precedentemente assegnate mettendole in relazione con quelle della gliceraldeide risultarono corrispondere alle loro vere configurazioni assolute, significando quindi che le configurazioni relative divennero anche assolute.[7]
Limiti della convenzione di Fischer-Rosanoff
La convenzione di Fischer-Rosanoff da luogo ad ambiguità quando viene applicata a molecole con più di un centro chirale. Se ad esempio si considera il D-(+)-glucosio, il sistema D-L ci da informazioni circa la configurazione del solo carbonio 2, quando nella molecola sono presenti altri tre centri asimmetrici, i carboni 3, 4 e 5.[3]
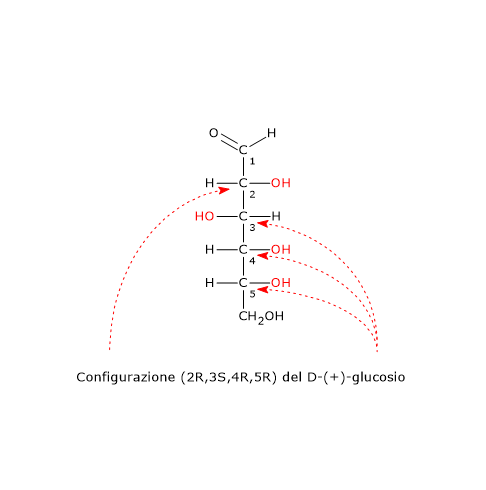
In questi casi il sistema RS, sviluppato nel 1956 da Robert Sidney Cahn, Christopher Ingold, e Vladimir Prelog, assegnando una configurazione a ogni singolo centro chirale permette di descrivere senza incertezze la stereochimica della molecola. Nel caso del D-(+)-glucosio si ha la configurazione (2R,3S,4R,5R).[5][8]
Va anche notato che, utilizzando la convezione di Fischer-Rosanoff, in base al centro chirale scelto come riferimento, una stessa molecola può appartenere sia alla serie D che a quella L.
Bibliografia
- ^ Rosanoff M.A. On Fischer’s classification of stereo-isomers. J Am Chem Soc 1906:28(1);114-121. doi:10.1021/ja01967a014
- ^ IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. doi:10.1351/goldbook
- ^ a b Garrett R.H., Grisham C.M. Biochemistry. 7th Edition. Cengage Learning, 2023.
- ^ a b Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
- ^ a b c Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
- ^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
- ^ Bijvoet J.M., Peerdeman A.F., Van Bommel A.J. Determination of the absolute configuration of optically active compounds by means of X-rays. Nature 1951;168(4268):271. doi:10.1038/168271a0
- ^ Cahn R.S., Ingold C., Prelog V. Specification of molecular chirality. Angew Chem 1966:5(4); 385-415. doi:10.1002/anie.196603851
Domande Frequenti
Cos'è la convenzione di Fischer-Rosanoff o sistema D-L?
È un sistema di nomenclatura stereochimica che classifica le molecole chirali, come carboidrati e amminoacidi, in due serie (D o L). La classificazione si basa sul confronto della loro configurazione relativa rispetto a quella della gliceraldeide, presa come standard di riferimento.
C'è legame tra configurazione D-L e rotazione ottica?
No, non esiste alcuna correlazione diretta. I prefissi D e L indicano esclusivamente la disposizione spaziale dei gruppi rispetto alla gliceraldeide. La direzione della rotazione della luce polarizzata è una proprietà fisica indicata separatamente con i segni (+) o (-).
Come si assegna la serie D o L a zuccheri e amminoacidi?
Nei carboidrati si considera la configurazione del carbonio chirale più distante dal gruppo carbonilico. Negli α-amminoacidi si valuta la disposizione dei quattro gruppi legati al carbonio asimmetrico α, dove in natura prevale nettamente la configurazione stereochimica L.
Quali sono i limiti principali della convenzione di Fischer?
Il sistema D-L genera ambiguità nelle molecole con più centri stereogenici, poiché definisce la configurazione globale basandosi su un solo carbonio di riferimento. Per mappare ogni centro chirale in modo assoluto e univoco, nel 1956 è stato introdotto il sistema RS (CIP).