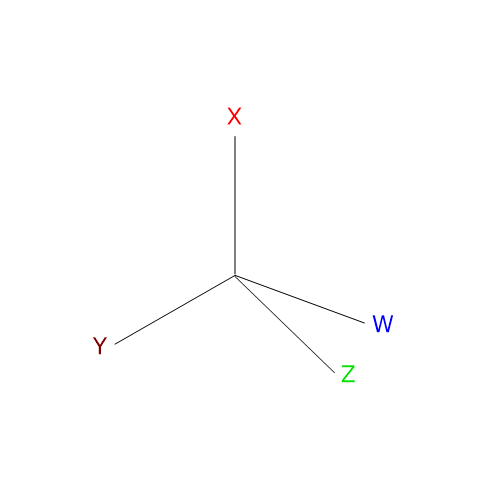
Chirality is the geometric property of a group of points or atoms in space, or of a solid object, of not being superimposable on its mirror image.[1] These structures, defined as chiral, have the peculiar characteristic of lacking second-order symmetry elements, namely a plane of symmetry, a center of inversion, or a rotation-reflection axis.[2]
The term chirality, from the Greek cheir meaning “hand”, was coined by Lord Kelvin, who introduced it during the “Baltimore Lectures”, a series of talks held at Johns Hopkins University in Baltimore, starting on October 1st, 1884, and published twenty years later, in 1904.[3]
The world is rich in chiral objects: your hands are the prime example, but many others exist, from the shell of a snail to a spiral galaxy. In chemistry, especially in organic chemistry, chirality is a property of primary importance, because molecules such as carbohydrates, many amino acids, and numerous drugs are chiral.[4]
Chiral molecules can exist in two forms: mirror images of each other that are non-superimposable. In other words, no combination of rotations or translations in the plane of the sheet can align them perfectly. These molecules are called enantiomers, from the Greek enántios, meaning “opposite”, and meros, meaning “part”.[5]
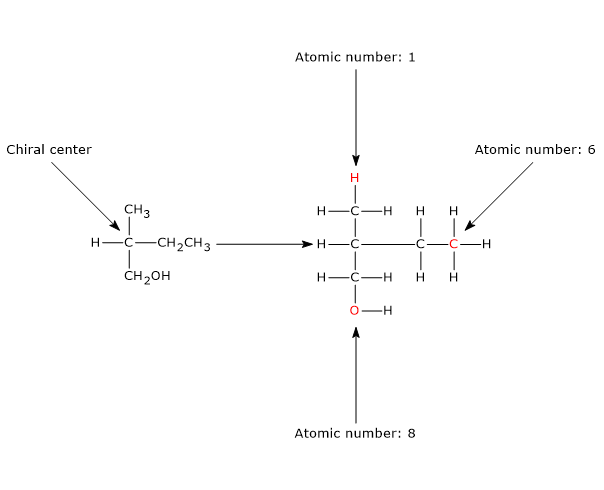
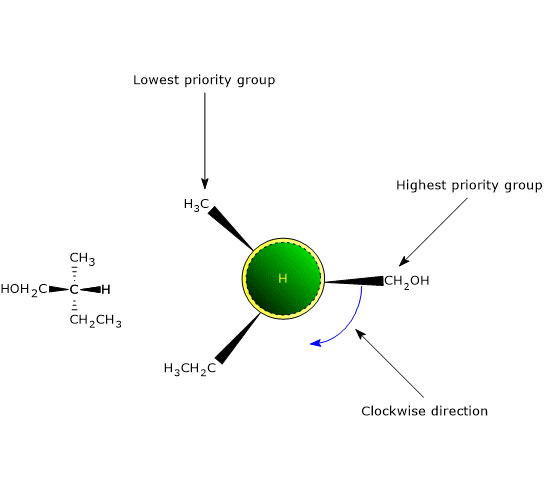
The most common cause of chirality in a molecule is the presence of a chiral center or chirality center, also called an asymmetric center, an atom bonded to a set of atoms or functional groups arranged in space such that the resulting molecule can exist as two enantiomers.
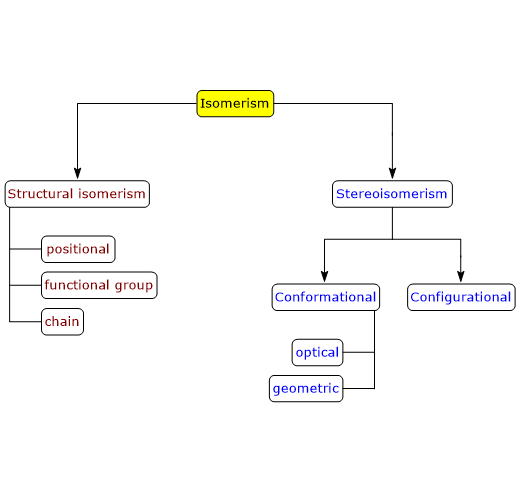
Enantiomers are a type of stereoisomers, which can be defined as isomers that have the same number and types of atoms and bonds, but differ in the spatial orientation of those atoms.[2]
Contents
Enantiomers
Two enantiomers of a chiral molecule, being non-superimposable, are distinct compounds. But how do they differ?
Each pair of enantiomers has identical physical and chemical properties with respect to achiral characteristics, such as melting point, boiling point, refractive index, infrared spectrum, solubility in the same solvent, and reaction rate with achiral reagents.
The differences emerge when enantiomers interact with chemical or physical phenomena that possess chirality.
- From a chemical point of view, two enantiomers can be distinguished by how they interact with chiral structures, such as the binding site of a chiral receptor or the active site of a chiral enzyme.
- From a physical point of view, they differ in their interaction with plane-polarized light, which exhibits chiral properties, in other words, they display optical activity.[6]
Chirality and optical activity
The optical activity of materials such as quartz and, more importantly, of organic compounds such as sugars or tartaric acid, was discovered in 1815 by the French scientist Jean-Baptiste Biot.[7]
Chiral molecules can be classified based on the direction in which plane-polarized light is rotated, from the observer’s point of view, when it passes through a solution containing them.
- If a solution containing one enantiomer rotates plane-polarized light in a clockwise direction, the molecule is called dextrorotatory or dextrorotary, from the Latin dexter, meaning “right”, and is designated by the prefixes (+)- or d-, from dextro-.
- If the solution rotates plane-polarized light in a counterclockwise direction, the molecule is called levorotatory or levorotary, from the Latin laevus, meaning “left”, and is designated by the prefixes (–)- or l-, from laevo-.[8]
Obviously, if we consider a pair of enantiomers, one is dextrorotatory and the other levorotatory.
Currently, it is not possible to reliably predict the magnitude, direction, or sign of the optical rotation caused by an enantiomer. Conversely, the optical activity of a molecule provides no information about the spatial arrangement of the chemical groups attached to its chirality center.
Note: a system containing molecules that all have the same chirality (i.e., the same “handedness”) is called enantiomerically pure or enantiopure.[9]
Pasteur and the discovery of enantiomers
In 1848, thirty-three years after Biot’s work, studies on the optical activity of molecules led Louis Pasteur, who had been a student of Biot, to observe that, following the recrystallization of a concentrated aqueous solution of sodium ammonium tartrate (which was optically inactive), two types of crystals precipitated that were non-superimposable mirror images of each other.[10]
After separating them with tweezers, Pasteur discovered that the solutions obtained by dissolving equimolar amounts of the two types of crystals were optically active. Interestingly, the rotation angle of plane-polarized light was equal in magnitude but opposite in direction.
Since the differences in optical activity were due to the dissolved sodium ammonium tartrate crystals, Pasteur hypothesized that the molecules themselves must also be non-superimposable mirror images of each other, just like the crystals. These were what we now call enantiomers.[11]
It was Pasteur who first used the term asymmetry to describe this property, which would later be termed chirality by Lord Kelvin.[12]
Racemic mixtures
A solution containing equal amounts of each member of a pair of enantiomers is called a racemic mixture or racemate. These solutions are optically inactive: there is no net rotation of plane-polarized light, since the quantities of dextrorotatory and levorotatory molecules are exactly the same.[13]
Unlike what occurs in biochemical processes, the chemical synthesis of chiral molecules that does not involve chiral reactants, or is not followed by enantiomer separation methods, inevitably leads to the production of a racemic mixture.[9]
Pharmaceutical chemistry is among the fields most affected by this. As previously mentioned, two enantiomers are distinct compounds. Many chiral drugs are synthesized as racemic mixtures; however, the desired pharmacological activity often resides in only one enantiomer, called the eutomer, while the other, known as the distomer, is less active or inactive.[14]
An example is ibuprofen, an arylpropionic acid derivative and anti-inflammatory drug: only the S-enantiomer, named according to the nomenclature system called RS system, exhibits pharmacological activity.
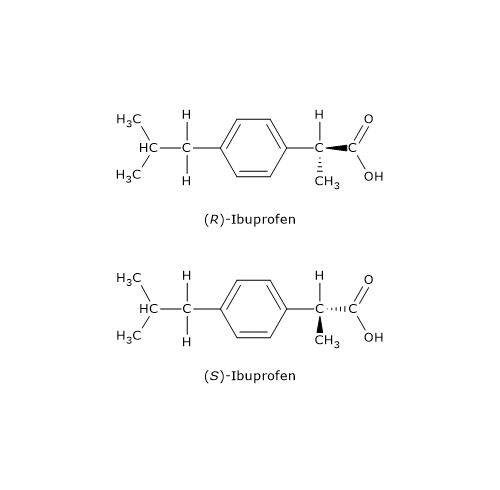
Arylpropionic acid derivatives are commonly sold as racemic mixtures. A racemase enzyme in the liver converts the distomer into the eutomer.[15]
However, in some cases, the distomer may cause harmful effects and must therefore be removed from the racemic mixture. A tragic example is thalidomide, a sedative and anti-nausea drug sold as a racemic mixture from the 1950s until 1961, and also taken during pregnancy.
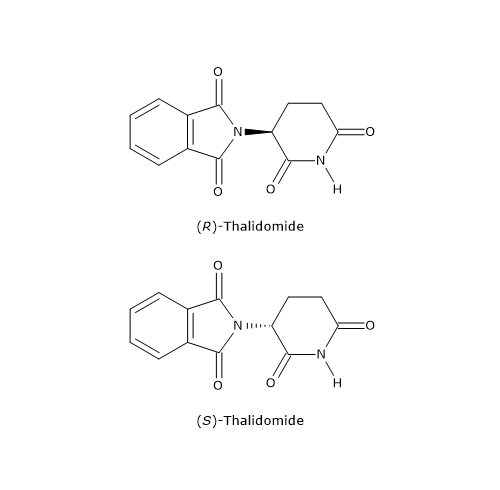
The distomer, the S-enantiomer, was found to cause serious birth defects, particularly phocomelia (malformation of limbs). This remains one of the most striking examples of the importance of the chiral properties of molecules, and it prompted health organizations to encourage the pharmaceutical industry to develop drugs, thalidomide included, containing only a single enantiomer.[16]
Chirality centers
Any tetrahedral atom bearing four different substituents can function as a chirality center.
The classic example is the carbon atom, but other group IVA elements in the periodic table, such as the semimetals silicon and germanium, also adopt tetrahedral geometry and can serve as chiral centers.[2]
Another notable example is the phosphorus atom in organic phosphate esters: when it adopts a tetrahedral arrangement and is bonded to four different substituents, it too becomes a chirality center.[4]
The nitrogen atom in a tertiary amine, an amine in which nitrogen is bonded to three different groups, can also be a chiral center. In such compounds, nitrogen occupies the center of a tetrahedron, with its four sp3 hybrid orbitals directed toward the vertices: three are occupied by substituents, while the fourth contains a nonbonding electron pair.
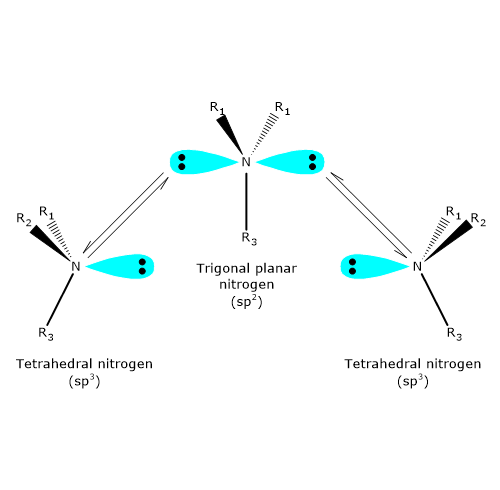
At room temperature, however, nitrogen undergoes rapid inversion of configuration. This process, known as nitrogen inversion, involves a fast oscillation in which the nitrogen atom passes through a planar, sp2 hybridized transition state. As a result, if the nitrogen atom is the only chirality center in a molecule, the compound exhibits no optical activity, because it effectively exists as a racemic mixture.Nitrogen inversion does not occur only in specific cases, such as when nitrogen is part of a cyclic structure that prevents this motion. Therefore, the mere presence of a chirality center may not always be sufficient to allow for the isolation or resolution of enantiomers.[4]
Note: in 1874, Jacobus Henricus van ‘t Hoff and Joseph Achille Le Bel, building on Pasteur’s work, were the first to propose the theory of the tetrahedral carbon atom. For this pioneering contribution, van ‘t Hoff was awarded the first Nobel Prize in Chemistry in 1901.[17]
Chirality in the absence of a chiral center
Chirality can also arise in the absence of a traditional chiral center, often due to the restricted rotation around certain bonds, typically a double bond or a hindered single bond.[18] This phenomenon is observed in cases such as:
- allene derivatives: organic compounds containing two cumulated double bonds, i.e., two double bonds located on the same carbon atom;[19]
- biphenyl derivatives: compounds consisting of two aromatic rings connected by a single bond that cannot freely rotate due to steric hindrance from substituents.[20]
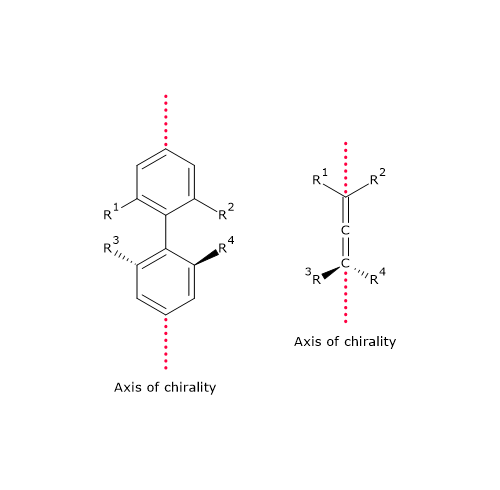 In such cases, chirality results from the presence of an axis of chirality rather than a center. This form of chirality is known as axial chirality.[21]
In such cases, chirality results from the presence of an axis of chirality rather than a center. This form of chirality is known as axial chirality.[21]
Meso compounds
Meso compounds are stereoisomers that contain two or more chiral centers but are superimposable on their mirror image. As such, they are achiral and therefore optically inactive. A defining feature of meso compounds is the presence of an internal mirror plane that bisects the molecule, with each half being the mirror image of the other.[8]
Meso compounds are classified as diastereomers, meaning stereoisomers that are not enantiomers.
For a molecule with n chirality centers, the maximum number of possible stereoisomers is 2n.[2]
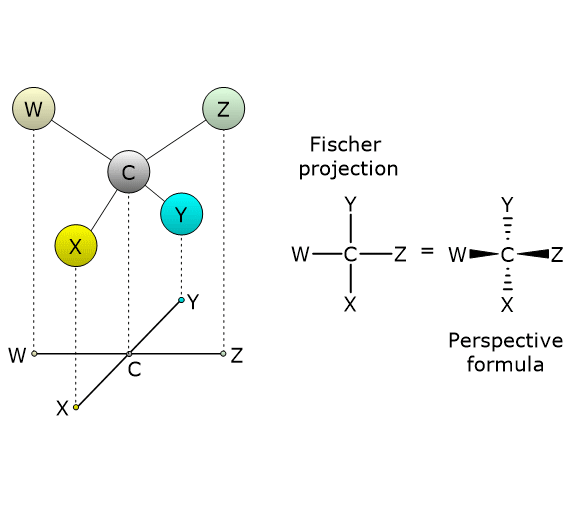
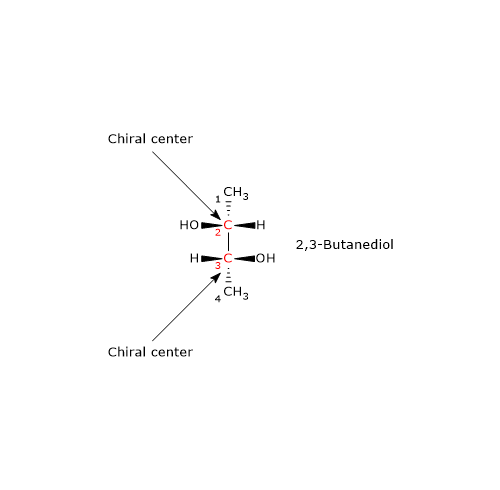
Consider 2,3-butanediol, which contains two chirality centers, specifically at carbon atoms 2 and 3. According to the formula, there are 22 = 4 possible stereoisomers, whose structures are depicted in the accompanying figure as Fischer projections, labeled A, B, C, and D.
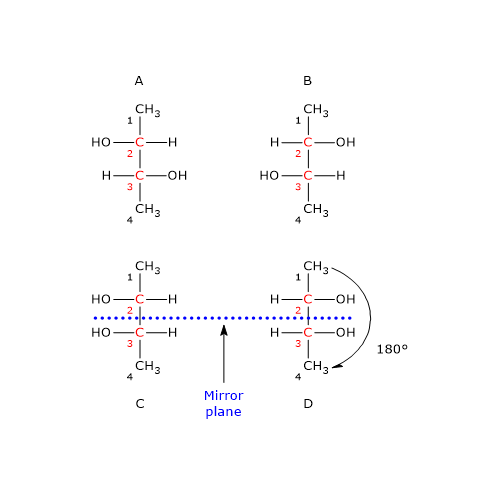
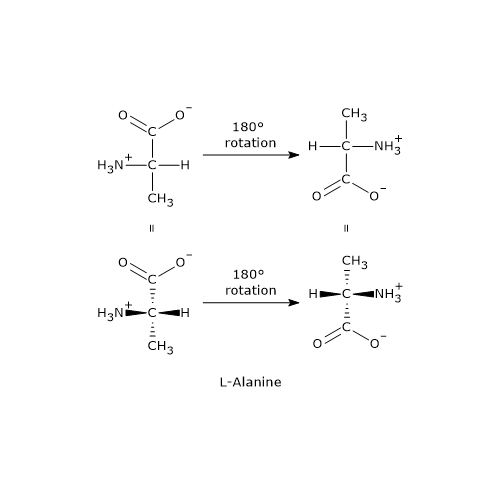
- Structures A and B are non-superimposable mirror images of each other, and thus constitute a pair of enantiomers.
- Structures C and D are also mirror images of each other, but they are superimposable. In fact, if either structure is rotated by 180 degrees, it aligns perfectly with the other. Therefore, C and D are not enantiomers; they are the same compound in different orientations.
Furthermore, these structures possess an internal mirror plane that divides the molecule into two symmetric halves, each a mirror image of the other. Structure C (or D) is therefore a meso compound: it contains chiral centers, is superimposable on its mirror image, and has an internal plane of symmetry that results in two mirror-image halves.[2]
References
- ^ Chirality, in IUPAC Compendium of Chemical Terminology, 5th ed. International Union of Pure and Applied Chemistry; 2025. Online version 5.0.0, 2025. doi:10.1351/goldbook.C01058
- ^ a b c d e Solomons T.W.G., Fryhle C.B., Snyder S.A. Solomons’ organic chemistry. 12th Edition. John Wiley & Sons Incorporated, 2017.
- ^ Kelvin W.T. Baltimore lectures on molecular dynamics and the wave theory of light. Clay C. J., London: 1904:619. https://archive.org/details/baltimorelecture00kelviala/mode/2
- ^ a b c Morsch L., Farmer S., Cunningham K., Sharrett Z., Shea K.M. Organic Chemistry II. Open Educational Resources: Textbooks, Smith College, Northampton, MA, 2023. https://scholarworks.smith.edu/textbooks/6
- ^ Enantiomer, in IUPAC Compendium of Chemical Terminology, 5th ed. International Union of Pure and Applied Chemistry; 2025. Online version 5.0.0, 2025. doi:10.1351/goldbook.E02069
- ^ Gogoi A., Konwer S., Zhuo G.Y. Polarimetric measurements of surface chirality based on linear and nonlinear light scattering. Front Chem 2021;8:611833. doi:10.3389/fchem.2020.611833
- ^ Flack H.D. Louis Pasteur’s discovery of molecular chirality and spontaneous resolution in 1848, together with a complete review of his crystallographic and chemical work. Acta Crystallogr A 2009;65(Pt 5):371-89. doi:10.1107/S0108767309024088
- ^ a b Soderberg T. Organic chemistry with a biological emphasis. Volume I. Chemistry Publications. 2019. https://digitalcommons.morris.umn.edu/chem_facpubs/1
- ^ a b Ariëns E.J. Stereochemistry: a source of problems in medicinal chemistry. Med Res Rev 1986;6(4):451-66. doi:10.1002/med.2610060404
- ^ Vantomme G., Crassous J. Pasteur and chirality: a story of how serendipity favors the prepared minds. Chirality 2021;33(10):597-601. doi:10.1002/chir.23349
- ^ Geison G.L. The private science of Louis Pasteur. Princeton University Press, 2014.
- ^ Pasteur L. Memoires sur la relation qui peut exister entre la forme crystalline et al composition chimique, et sur la cause de la polarization rotatoire. C R Acad Sci 1848;26:535‐538.
- ^ Racemate, in IUPAC Compendium of Chemical Terminology, 5th ed. International Union of Pure and Applied Chemistry; 2025. Online version 5.0.0, 2025. 10.1351/goldbook.R05025
- ^ Tamatam R., Shin D. Asymmetric synthesis of US-FDA approved drugs over five years (2016-2020): a recapitulation of chirality. Pharmaceuticals (Basel) 2023;16(3):339. doi:10.3390/ph16030339
- ^ Evans A.M., Nation R.L., Sansom L.N., Bochner F., Somogyi A.A. The relationship between the pharmacokinetics of ibuprofen enantiomers and the dose of racemic ibuprofen in humans. Biopharm Drug Dispos 1990;11(6):507-18. doi:10.1002/bdd.2510110605
- ^ Tokunaga E., Yamamoto T., Ito E., Shibata N. Understanding the thalidomide chirality in biological processes by the self-disproportionation of enantiomers. Sci Rep 2018;8(1):17131. doi:10.1038/s41598-018-35457-6
- ^ Jacobus H. van ‘t Hoff – Facts. NobelPrize.org. Nobel Prize Outreach 2025. Sun. 1 Jun 2025. https://www.nobelprize.org/prizes/chemistry/1901/hoff/facts/
- ^ Capozziello S., Lattanzi A. Geometrical approach to central molecular chirality: a chirality selection rule. Chirality 2003;15:227-230. doi:10.1002/chir.10191
- ^ Ōki M. The chemistry of rotational isomers. New York: Springer; 1993.
- ^ Runge W. The chemistry of the allenes. Vol. 2, Landor S R: Academic Press; 1982.
- ^ Axial chirality, in IUPAC Compendium of Chemical Terminology, 5th ed. International Union of Pure and Applied Chemistry; 2025. Online version 5.0.0, 2025. doi:10.1351/goldbook.A00547
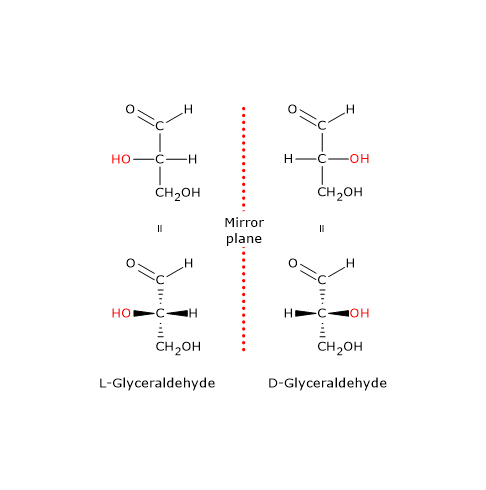
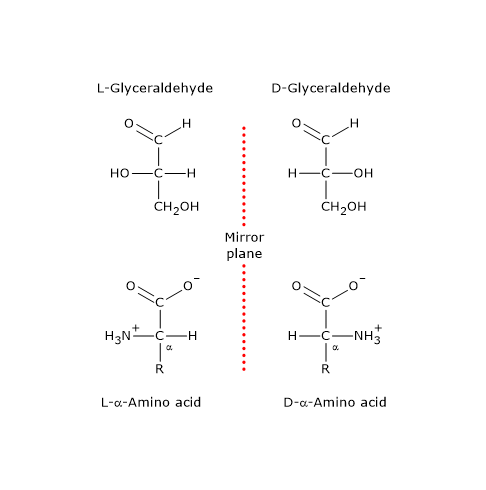
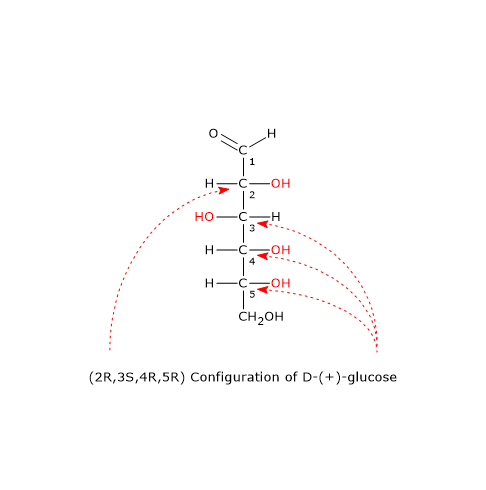
 Note: in nature, D-sugars are far more common than L-sugars.
Note: in nature, D-sugars are far more common than L-sugars.