The pyruvate dehydrogenase complex (PDC) is one of the mitochondrial multienzyme complexes, and is composed of three different enzymes:
- pyruvate dehydrogenase or E1 (EC 1.2.4.1);
- dihydrolipoyl transacetylase or dihydrolipoamide acetyltransferase or E2 (EC 2.3.1.12);
- dihydrolipoyl dehydrogenase or dihydrolipoamide dehydrogenase or E3 (EC 1.8.1.4).
Each of these enzymes is present in multiple copies whose number, and then the size of the complex itself, varies from species to species, with molecular masses ranging from 4×106 to 1×107 daltons.
The multienzyme complex contains additional subunits:
- five different coenzymes;
- in plants, fungi, and among animals, aves and mammals, two enzymes with regulatory properties: a Mg2+-dependent pyruvate dehydrogenase kinase (EC 2.7.1.99) and a Ca2+-activated pyruvate dehydrogenase phosphatase(EC 3.1.3.43);
- in eukaryotes, a binding protein called E3BP.
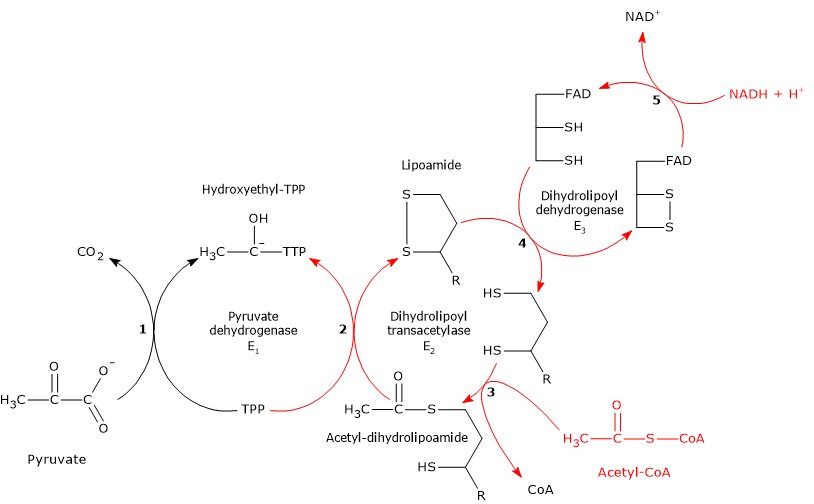
The pyruvate dehydrogenase complex catalyzes, through five sequential reactions, the oxidative decarboxylation of pyruvate, the conjugate base of pyruvic acid, to form a carbon dioxide molecule (CO2) and the acetyl group of acetyl-coenzyme A or acetyl-CoA, with the release of two electrons, carried by NAD.
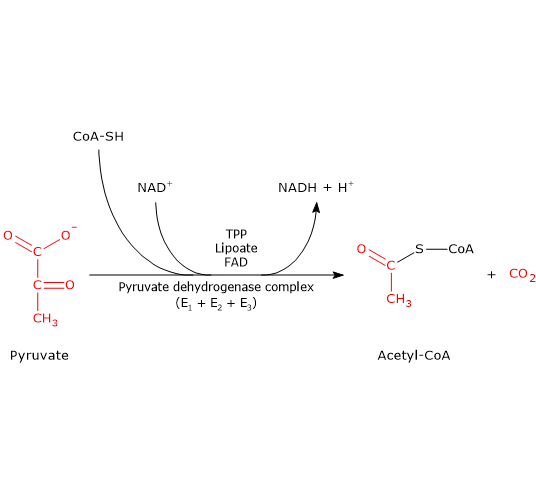
The overall reaction is essentially irreversible, with a ΔG°’ of -8.0 kcal/mol (-33.4 kJ/mol), and requires the intervention of the three enzymes, whose activities are sequentially coordinated. During the reactions, the intermediate products remain bound to the enzymes and, at the end of the reaction sequence, the multienzyme complex is ready for the next cycle.
Note: the pyruvate dehydrogenase complex catalyzes the same reactions through similar mechanisms in all organisms.
Contents
- Coenzymes of the pyruvate dehydrogenase complex
- Where is the pyruvate dehydrogenase complex located?
- Functions of the pyruvate dehydrogenase complex
- Mitochondrial pyruvate transport
- Structure of the pyruvate dehydrogenase complex
- Reaction of pyruvate dehydrogenase
- Regulation of the pyruvate dehydrogenase complex
- References
Coenzymes of the pyruvate dehydrogenase complex
Five coenzymes are used in the pyruvate dehydrogenase complex reactions. They are thiamine pyrophosphate (TPP), flavin adenine dinucleotide (FAD), coenzyme A (CoA), nicotinamide adenine dinucleotide (NAD), and lipoic acid.
- Thiamine pyrophosphate is the active form of thiamine or vitamin B1. TPP is the coenzyme of pyruvate dehydrogenase, to which it is strictly bound through noncovalent interactions. It is involved in the transfer of hydroxyethyl or “activated aldehyde” groups.
- Flavin adenine dinucleotide is one the active forms of riboflavin or vitamin B2; the other is flavin mononucleotide (FMN).
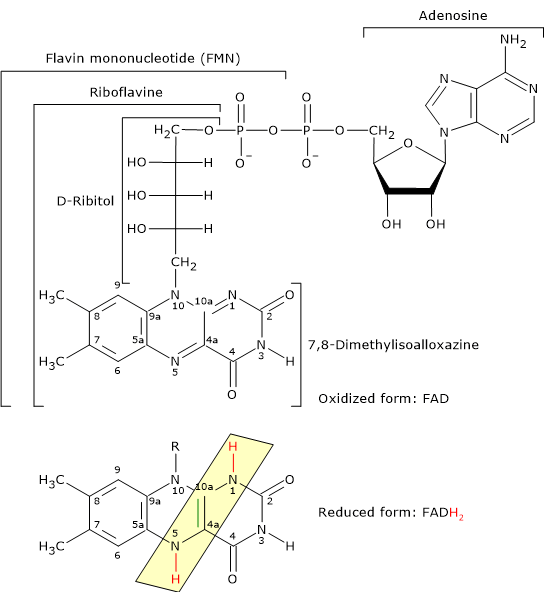
Reduced and Oxidized Form of Flavin Adenine Dinucleotide FAD is the coenzyme of dihydrolipoyl dehydrogenase, to which it is strictly bound. Like NAD, it participates in electron transfer, or hydride ion (:H– or H+ + 2e–) transfer.
- Coenzyme A consists of a β-mercaptoethylamine group connected to pantothenic acid or vitamin B5 through an amide linkage, which, in turn, is bonded to 3′-phosphoadenosine moiety, through a pyrophosphate bridge.
CoA is involved in the reaction catalyzed by dihydrolipoyl transacetylase, and acts as a carrier of acyl groups.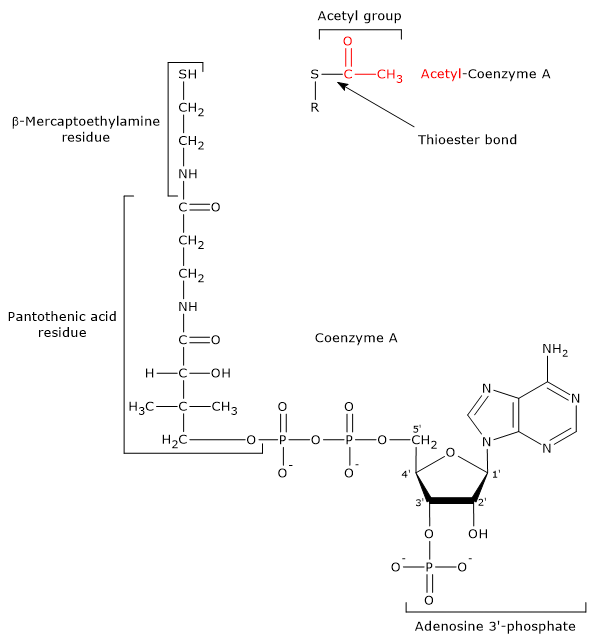
Coenzyme A and Acetyl-coA The β-mercaptoethylamine moiety terminates with a sulfhydryl group (–SH), a reactive thiol crucial for the role played by the coenzyme, because the acyl groups bonded to it through a thioester bond have a high standard free energy of hydrolysis. This provides acyl groups with a high transfer potential, equal to -31.5 kcal/mol (-7.5 kJ/mol), slightly more exergonic [1 kJ/mol (0.2 kcal/mol )] than that for the hydrolysis of ATP to ADP and Pi. Thioesters have therefore a high transfer potential of the acyl group and can donate it to a variety of molecules, that is, such acyl group can be considered as an activated group ready for a transfer. It is also possible to state that the formation of the thioester bond allows to conserve a portion of the free energy derived from the oxidation of the metabolic fuel. It should be noted that coenzyme A is also abbreviated as CoA-SH to emphasize the role played by the thiol group.
Note: in the thioester bond a sulfur atom sits in the position where an oxygen atom is in the ester bond.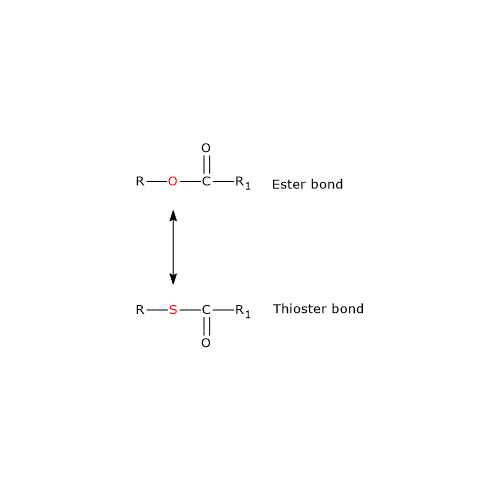
Ester and Thioester Bonds
- Nicotinamide adenine dinucleotide can be synthesized from tryptophan, an essential amino acid, or from niacin or vitamin B3 or vitamin PP, from Pellagra-Preventing, the source of the nicotinamide moiety.
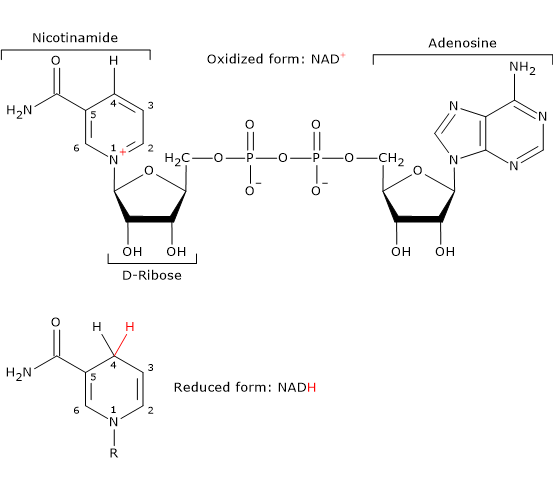
NAD is involved in the reaction catalyzed dihydrolipoyl dehydrogenase, and, like FAD, participates in electron transfer, or hydride transfer.
- Unlike the other coenzymes of the pyruvate dehydrogenase complex, lipoic acid does not derive, directly or indirectly, from vitamins and/or essential amino acids, that is, from building blocks that cannot be synthesized de novo by the organism and must be supplied by the diet.
It is the coenzyme of dihydrolipoyl transacetylase, to which it is covalently bound, through an amide linkage, to the ɛ-amino group of a lysine residue to form a lipoyl-lysine or lipoamide, the so-called lipoyllysyl arm. It couples electron transfer to acyl group transfer.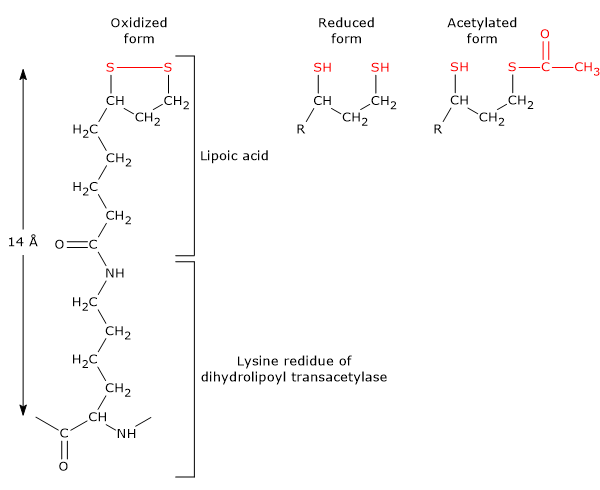
Lipoamide or Lipoyl-lysine Lipoic acid has two thiol groups that can undergo a reversible intramolecular oxidation to form a disulfide bridge (-S-S-), a reaction analogous to that between two cysteine (Cys) residues of a protein.
Because the disulfide bridge (note: a cyclic disulfide) is capable of undergoing redox reactions, during the reactions catalyzed by the pyruvate dehydrogenase complex, it is first reduced to dihydrolipoamide, a dithiol or the reduced form of the prosthetic group, and then, reoxidized to the cyclic form.
Note
Many enzymes require small non protein components, called cofactors, for their catalytic activity. Cofactors can be metal ions or small organic or metalloorganic molecules, and are classified as coenzymes and prosthetic groups.
A prosthetic group is cofactor that binds tightly to an enzyme by non-covalent or covalent bond, namely, it is permanently bound to the protein.
A coenzyme is cofactor that is not permanently bound to the enzyme.
Where is the pyruvate dehydrogenase complex located?
In eukaryotes, the pyruvate dehydrogenase complex, like the enzymes for citric acid cycle and oxidation of fatty acids, is located in the mitochondrion, where is associated with the surface of the inner membrane facing the matrix.
In prokaryotes, it is located in the cytosol.
Functions of the pyruvate dehydrogenase complex
The main functions of the pyruvate dehydrogenase complex are to produce acetyl-CoA and NADH.
- The acetyl group linked to coenzyme A, an activated acetate, depending on the metabolic conditions within the cell and/or the cell type, can be:
oxidized to two carbon dioxide molecules via the citric acid cycle reactions to harvest a portion of the potential energy stored in the form of ATP or GTP;
utilized for the synthesis of fatty acids, cholesterol, steroids, isoprenoids, ketone bodies and acetylcholine.
It is therefore possible to state that, depending on the metabolic conditions and/or cell type, the pyruvate dehydrogenase complex commits carbon intermediates from amino acid and glucose catabolism to:
citric acid cycle, and then to the production of energy, e.g. in skeletal muscle in aerobic conditions, and, always, in cardiac muscle;
synthesis of lipids and acetylcholine.
- In aerobic organisms, NADH can be oxidized to NAD+ via hydride ion transfer to the mitochondrial electron transport chain that, in turn, carries the two electrons to molecular oxygen (O2), allowing the production of 2.5 ATP molecules per pair of electrons.Note: in anaerobic organisms there are electron acceptors alternative to oxygen, such as sulfate or nitrate.
Conceptually, the pyruvate dehydrogenase complex is the bridge between glycolysis and the citric acid cycle. However, due to the irreversibility of the overall reaction catalyzed by the multienzyme complex, it acts as an one-way bridge: pyruvate can be decarboxylated, oxidized and the remaining acetyl unit linked to the CoA, but it is not possible to carry out the opposite reaction, namely, to convert acetyl-CoA into pyruvate.
The irreversibility of this reaction, and the absence of alternative pathways, explain why it is not possible to use acetyl-CoA, and therefore fatty acids, as a substrate for gluconeogenesis.
Other sources of acetyl-CoA
Other than pyruvate, the acetyl group of acetyl-CoA can derive from the oxidation of fatty acids and the catabolism of many amino acids. However, regardless of its origin, acetyl-CoA represents an entry compound for new carbon units into the citric acid cycle. And, it is also possible to state that the acetyl group of acetyl-CoA represents the form in which most of the carbon enters the cycle.
Mitochondrial pyruvate transport
In eukaryotes, glycolysis occurs in the cytosol whereas all the subsequent steps of the aerobic metabolism, that is, the reactions catalyzed by the pyruvate dehydrogenase complex, the citric acid cycle, the electron transport chain, and the oxidative phosphorylation occur in the mitochondria.
Similarly to most other metabolites and anions, the transport of pyruvate across the outer mitochondrial membrane is probably mediated by a relatively non-specific, voltage-dependent anion channel. Conversely, its transport across the inner mitochondrial membrane occurs through a specific transporter made up of two proteins named MPC1 and MPC2, acronym of mitochondrial pyruvate carrier, which form a hetero-oligomeric complex in the membrane.
Structure of the pyruvate dehydrogenase complex
Although the pyruvate dehydrogenase complex is composed of multiple copies of three different enzymes and catalyzes the same reactions by similar mechanisms in all the organisms in which it is present, it has a very different quaternary structure.
The structure of E. coli multienzyme complex, which has a weight of ∼4,600 kD and a diameter of ∼300 Å, was the first to be characterized, thanks to the work of Lester Reed. In this complex, 24 units of dihydrolipoyl transacetylase form a structure with cubic symmetry, namely, the enzymes, associated as trimers, are placed at the corners of a cube. Dimers of pyruvate dehydrogenase are associated with dihydrolipoyl transacetylase core, at the center of each edge of the cube, for a total of 24 units. Finally, dimers of dihydrolipoyl dehydrogenase are located at the center of each of the six faces of the cube, for a total of 12 units. Note that the entire complex is composed of 60 units. A similar structure with cubic symmetry is also found in most other Gram-negative bacteria.
In some Gram-positive bacteria and in eukaryotes, the pyruvate dehydrogenase complex has a dodecahedral form, namely, that of a regular polyhedron with 20 vertices, 12 pentagonal faces, and 30 edges, with icosahedral symmetry, also called I symmetry. Considering for example the multienzyme complex present in mitochondria, it is the largest known multienzyme complex, with a weight of ∼10,000 kD and a diameter of ∼500 Å, so more than 5 times the size of a ribosome. Noteworthy, it can be visualized with the electron microscope.
The complex is composed of a dodecahedral core, with a diameter of about 25 nm, formed, as in Gram-negative bacteria, by dihydrolipoyl transacetylase, but consisting of 20 trimers of the enzyme, for a total of 60 units, located at the vertices of the structure. Its core is surrounded by 30 units of pyruvate dehydrogenase, one centered on each edge, and 12 units of dihydrolipoyl dehydrogenase, one centered on each face. The entire complex is therefore composed of 102 units.
The quaternary structure of the complex is further complicated, as mentioned previously, by the presence of three additional subunits: a pyruvate dehydrogenase kinase, a pyruvate dehydrogenase phosphatase, and the E3-binding protein.
Kinase and phosphatase are bound to the dihydrolipoyl transacetylase core.
E3BP is bound to each of the 12 pentagonal faces, and therefore is present in about 12 copies. It is required to bind dihydrolipoyl dehydrogenase to the core of dihydrolipoyl transacetylase, as demonstrated by the fact that its partial proteolysis decreases the binding ability of the dehydrogenase. In E3-binding protein it is possible to identify a C-terminal domain, that has no catalytic activity, and a lipoamide-containing domain, similar to that of dihydrolipoyl transacetylase, capable of accepting an acetyl group, too. However, the removal of this domain does not cause any reduction of the catalytic activity of the multienzyme complex.
Structure of pyruvate dehydrogenase
Pyruvate dehydrogenase of eukaryotes and some Gram-positive bacteria is composed of two different polypeptide chains, called α and β, associated to form a 2-fold symmetric α2β2 heterotetramer. Conversely, in E. coli and other Gram-negative bacteria the two subunits are fused to form a single polypeptide chain, and the enzyme is a homodimer.
The enzyme has two active sites.
Considering the heterotetrameric structure of Bacillus stearothermophilus pyruvate dehydrogenase, a Gram-positive bacteria, each thiamine pyrophosphate binds between the N-terminal domains of an α and a β subunit, at the end of a ∼21 Å deep funnel-shaped channel leading to the active site, with its reactive group, the thiazole ring, closest to the channel entrance. At the entrance this channel there are also two conserved loops, essential both for the catalytic activity of the enzyme and for its regulation.
The X-ray analysis of B. stearothermophilus enzyme, when it binds both TPP and the peripheral subunit-binding domain (PSBD) of dihydrolipoyl transacetylase, which binds to the C-terminal domain of the β subunits, has revealed that, in addition to a heterotetramer with a very tight structure, the two active sites have a different structure, in particular regarding the arrangement of the two conserved loops. In fact, in one enzyme subunit, in the presence of the activated form of thiamine pyrophosphate, the inner loop is ordered in a way that it blocks the entrance to the active site, whereas the loop at the entrance of the other active site is disordered and does not block the entrance.
This explains, from a structural point of view, the observed differences in the rate of substrate binding exhibited by the two active sites. A similar arrangement and asymmetry have been observed in all thiamine pyrophosphate-dependent enzymes of which the structure has been solved.
In addition to TPP and a magnesium ion (Mg2+), located in each of the two active sites, a third Mg2+ is located at the centre of the tetramer, within a ∼20 Å deep solvent-filled tunnel that connects the two active sites. The tunnel is largely lined by 10 conserved amino acid residues from all four subunits, in particular glutamate (Glu) and aspartate (Asp), six and four, respectively, plus other acidic residues around the TPP aminopyrimidine ring. And it should be underlined the absence of basic residues to neutralize them. Similar tunnels have been found in all thiamine pyrophosphate-dependent enzymes with known crystalline structure, with dimeric or tetrameric structure, for example in transketolase, an enzyme of the pentose phosphate pathway.
Note: B. stearothermophilus belongs to the phylum Firmicutes and is recently renamed Geobacillus stearothermophilus.
What is the function of the acidic tunnel?
Through mutagenesis experiments on B. stearothermophilus pyruvate dehydrogenase, the tunnel has been shown to play a role in the catalytic mechanism.
The change of some of the aforementioned acidic residues to neutral amino acids does not alter, compared with the wild-type pyruvate dehydrogenase:
- the efficiency of incorporation of the modified enzyme into the multienzyme complex;
- the structure of active sites;
- the quaternary structure of the enzyme.
However, rate of decarboxylation is reduced by over 70 percent compared to the wild-type enzyme, as well as, once the multienzyme complex is assembled with the mutant pyruvate dehydrogenase, the PDC activity, which is reduced by over 85 percent compared to the wild-type complex. But how does this occur?
Because the distance between the substituted amino acids and the active sites is ≥7 Å, that is, these amino acids are remote from the active sites of pyruvate dehydrogenase, they cannot directly influence its catalytic activity. Then, the catalytic mechanism described below was proposed.
Considering the apoenzyme, thiamine pyrophosphate binds fast and strongly to the first active site, is activated, and the active site is closed, thus protecting the zwitterionic thiazolium from the external environment.
Conversely, in the second active site TPP binds, but is not activated, and the active site remains in an open conformation.
In the first active site, pyruvate reacts with the thiazolium C-2, and thiamine pyrophosphate of the second active site, which is a general acid, donates a proton to the first site. The result is a decarboxylation in the first site and the activation of the coenzyme in the second site, which is then closed.
It should be noted that while the activation of the first thiamine pyrophosphate is the result of the binding to the active site, the activation of the second coenzyme, and therefore of the second active site, is coupled to the decarboxylation of pyruvate in the first active site. Or, from another point of view, while an active site requires a general acid, the other requires a general base.
Protons are needed for the catalytic activity, and their transport between the two active sites occurs via the acidic tunnel. They are reversibly shuttled along a chain of donor-acceptor groups provided by glutamate and aspartate residues and the entrained water, that act as a proton wire.
It seems, therefore, that unlike many other enzymes, in which the communication between the active sites occurs through conformational changes and subunit rearrangements, in pyruvate dehydrogenase and in the other TPP-dependent enzymes, the proton wire is the molecular basis of such communication.
At this point, the holoenzyme has been formed and the active sites are in a dynamic equilibrium, each exchanging between the dormant and the activated state. This seems to be the state in which the enzyme is found in vivo at the start of each catalytic cycle.
A consequence of such a mechanism is that, as the catalytic cycles occur, the two active sites are out of phase with each other, namely, when an active site requires a general acid, the other requires a general base, and vice versa.
Finally, it should be noted that this mechanism allows the switching of the loops that close the active sites so as to:
- coordinate substrate uptake and product release;
- explain the asymmetry existing between the two active sites.
Note: an apoenzyme is an enzyme that lacks the association of its cofactors. Conversely, an holoenzyme is an apoenzyme together with its cofactors. The apoenzyme is a catalytically inactive enzyme, whereas the holoenzyme is a catalytically active enzyme.
Structure of dihydrolipoyl transacetylase
Three functionally distinct domains can be identify in the structure of dihydrolipoyl transacetylase: an N-terminal lipoyl domain, a peripheral subunit-binding domain, and a C-terminal catalytic domain or acyltransferase domain. These domains are connected by 20- to 40 amino acid residues rich in alanine and proline, hydrophobic amino acids that are interspersed with charged residues. These linkers are highly flexible and largely extended, that allows the three domains to kept away from each other.
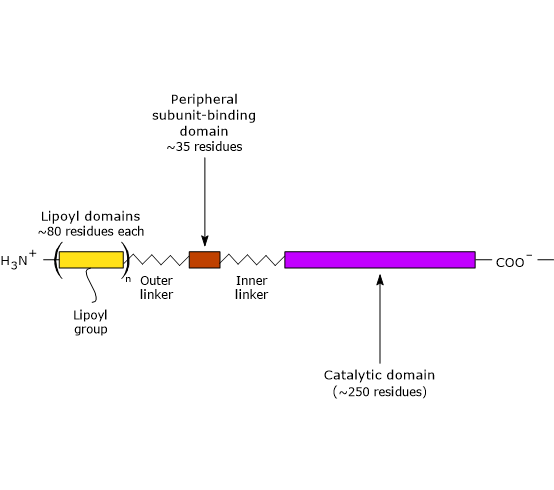
Note: flexible linkers are present in E3BP, too.
- The N-terminal lipoyl domain is composed of ∼80 amino acid residues, and is so called because it binds lipoic acid. The number of these domains depend on the species, ranging from one to three. For example, there is one domain in B. stearothermophilus and in yeasts, two in Streptococcus faecalis and in mammals, and three in Azotobacter vinelandii and E. coli.
The link between the ɛ-amino group of a lysine residue and lipoic acid leads to the formation of a flexible arm, the lipoyl-lysine, which has a maximum extended length of ∼14 Å. Adding the polypeptide segment which connects the N-terminal domain to the adjacent domain, whose length is greater than 140 Å, the resulting flexible tether is able to swings the lipoyl group between the active sites of pyruvate dehydrogenase and dihydrolipoyl dehydrogenase, as well as to interact with neighboring dihydrolipoyl transacetylases of the core.
It should be noted that the number of these tethers is 3 x 24 = 72 in E. coli, whereas in mammals 2 x 60 = 120, based on the number of N-terminal domains and the units of dihydrolipoyl transacetylase.
One pyruvate dehydrogenase can therefore acetylate numerous dihydrolipoyl transacetylases, and one dihydrolipoyl dehydrogenase can reoxidize many dihydrolipoamide groups.
Moreover, it also occurs:
an interchange of the acetyl groups between the lipoyl groups of the dihydrolipoyl transacetylase core;
the exchange of both acetyl groups and disulfides between the tethered arms.
- PSBD is composed of ∼35 amino acid residues arranged to form a globular structure that binds to both pyruvate dehydrogenase and dihydrolipoyl dehydrogenase, that is, it holds the multienzyme complex together.
- The C-terminal catalytic domain, which, of course, contains the active site, is composed of ∼250 amino acid residues arranged to form a hollow cage-like structure containing channels large enough to allow substrates and products to diffuse in and out. For example, CoA ad lipoamide, the two substrate of dihydrolipoyl transacetylase, bind, in their extended conformation, at the opposite ends of a channel located at the interface between each pair of subunits in each trimers.
Structure of dihydrolipoyl dehydrogenase
The structure of dihydrolipoyl dehydrogenase was deduced from studies of the enzyme in several microorganisms. It has a homodimeric structure, with each ∼470 amino acid residue chain folded into four domains, from the N-terminal to the C-terminal end: a FAD-binding domain, a NAD+-binding domain, a central domain, and an interface domain. All domains participate in the formation of the active site.
FAD is almost completely hidden inside the protein because, unlike thiol or NADH, it is easily oxidizable and must therefore be protected from the surrounding solution, namely, from O2. In fact, in the absence of the nicotinamide coenzyme, the phenol side chain of a tyrosine residue (Tyr), for example Tyr181 in the Gram-negative bacteria Pseudomonas putida, covers the NAD+-binding pocket so as to protect FADH2 from the contact with the surrounding solution.
Conversely, when NAD+ is located in the active site, the phenol side chain of the aforementioned tyrosine residue is interposed between the nicotinamide ring and the flavin ring.
In the active site of the enzyme’s oxidized form is also present a redox-active disulphide bridge. It forms between two cysteine residues located in a highly conserved segment of the polypeptide chain, e.g., Cys43 and Cys48 in P. putida, and is located on opposite side of the flavin ring with respect to the nicotinamide ring. The disulphide bridge links consecutive turns in a segment of a distorted α-helix, and, noteworthy, in the absence of such distortion, Cα atoms of the two cysteine residues would be too distant to allow the disulfide bridge to form.
Dihydrolipoyl dehydrogenase has therefore two electron acceptors: FAD and the redox-active disulphide bridge.
Note: the heterocyclic rings of NAD and FAD are parallel and in contact through van der Waals interactions; S48 is also in contact through van der Waals interactions with the flavin ring, on the opposite side of it from the NAD ring.
Reaction of pyruvate dehydrogenase
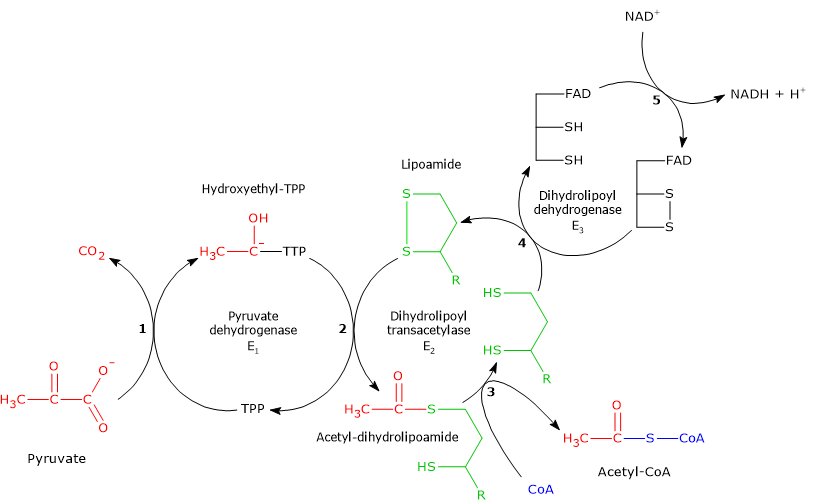
In the reaction sequence catalyzed by components of the pyruvate dehydrogenase complex, pyruvate dehydrogenase catalyzes the first two steps, namely:
- the decarboxylation of pyruvate to form CO2 and the hydroxyethyl-TPP intermediate;
- the reductive acetylation of the lipoyl group of dihydrolipoyl transacetylase.
The first reaction is essentially identical to pyruvate decarboxylase reaction (EC 4.1.1.1), which carries out a non-oxidative decarboxylation in glucose fermentation to ethanol. What differs is the fate of the hydroxyethyl group bound to thiamine pyrophosphate that, in the reaction catalyzed by pyruvate dehydrogenase is transferred to the next enzyme in the sequence, dihydrolipoyl transacetylase, whereas in the reaction catalyzed by pyruvate decarboxylase is converted into acetaldehyde.
Catalytic mechanism of pyruvate dehydrogenase
In thiamine pyrophosphate-dependent enzymes, the thiazolium ring is the active center, but only as dipolar carbanion or ylid, namely, as a dipolar ion, or zwitterion (German for “hybrid ion”), with positive charge on the N-3 and negative charge on C-2. Conversely, the positively charged thiazolium ring, that is, positive charged nitrogen and no charge on C-2, can be defined as “dormant” or inactive form.
The reaction begins with the nucleophilic attack by C-2 carbanion to the carbonyl carbon of pyruvate, which has the oxidation state of an aldehyde, and leads to the formation of a covalent bond between coenzyme and pyruvate.
Then, the cleavage of C-1–C-2 bond of pyruvate occurs. This leads to the release of the carboxyl group, namely, of the C-1 as CO2, while the remaining carbon atoms, C-2 and C-3, stay bound to the thiamine pyrophosphate as hydroxyethyl group. The cleavage of the C-1–C-2 bond, and therefore the decarboxylation of pyruvate, is favored by the fact that the negative charge on the C-2 carbon, that is unstable, is stabilized by the presence in the thiazolium ring of the positively charged N-3, a imine nitrogen (C=N+), that is, due to the presence of an electrophilic or electron deficient structure that acts as an electron sink or electron trap, in which the carbanion electrons can be delocalized by resonance.
At this point, the intermediate stabilized by resonance can be protonated to form hydroxyethyl-TPP.
Note: this first reaction catalyzed by pyruvate dehydrogenase is that in which the private dehydrogenase complex exercises its substratum specificity; furthermore, it is the slowest of the five reactions, hence limiting the rate of the overall reaction.
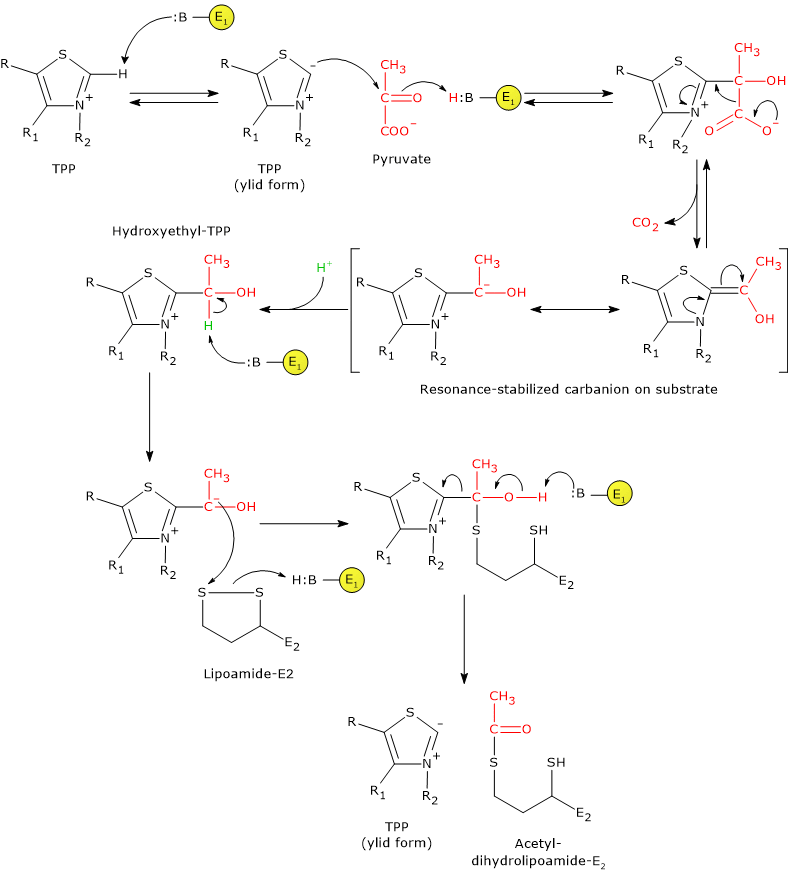
Pyruvate dehydrogenase then catalyzes the oxidation of the hydroxyethyl group to an acetyl group, and its transfer on lipoyllysyl arm of dihydrolipoyl transacetylase. The reaction begins with the formation of a carbanion on the hydroxylic carbon of the hydroxyethyl-TPP, by the removal of the carbon-linked proton by an enzyme base.
The carbanions carry out a nucleophilic attack on the lipoamide disulfide, with the formation of a high-energy acetyl-thioester bond with one of the two -SH groups. In this reaction, the oxidation of the hydroxyethyl group to an acetyl group occurs with the concomitant reduction of the lipoamide disulfide bond: the two electrons removed from the hydroxyethyl group are used to reduce the disulfide. This reaction is therefore a reductive acetylation accompanied by the regeneration of the active form of pyruvate dehydrogenase, namely, the enzyme with the thiazolium C-2 in the deprotonated form, the ylid or dipolar carbanion form.
Note that the energy derived from the oxidation of the hydroxyethyl group to an acetyl group drives the formation of the thioester bond between the acetyl group and coenzyme A.
Note: as previously said, the lipoyllysyl arm, arranged in an extended conformation in the channel where TPP is also found, allows the transfer of hydroxyethyl from hydroxyethyl-TPP to CoA, that is, it can move from the active site of pyruvate dehydrogenase to the active sites of dihydrolipoyl transacetylase, and then of dihydrolipoyl dehydrogenase.
A deeper look on thiamine pyrophosphate
Thiamine pyrophosphate molecule consists of three chemical moieties, from which its chemistry and enzymology depend: a thiazolium ring, a 4-aminopyrimidine ring, and the diphosphate side chain.
The diphosphate side chain binds the cofactor to the enzyme via the formation of electrostatic bonds between the negative charges carried by its phosphoryl groups and the positive charges carried by Ca2+ and Mg2+ ions, in turn, bound to highly conserved sequences, GlyAspGly (GDG) and GlyAspGly-X26-AsnAsn (GDG-X26-NN), respectively.
The thiazolium ring plays a central role in catalysis, due to its ability to form the C-2 carbanion, that is, a nucleophilic center on the C-2 atom.
Note: as mentioned previously in this article, once bound to the enzyme, thiamine pyrophosphate locates in the active site so that the thiazolium ring is positioned close to the channel entrance leading to the active site.
The aminopyrimidine ring has a dual function:
- it anchors the coenzyme holding it in place;
- it has a specific catalytic role, participating in acid/base catalysis, as evidenced by studies with thiamine pyrophosphate analogs in which each of the three nitrogen atoms of the ring were replaced in turn. These studies demonstrated that the N-1’ atom and the N-4’-amino group are required, whereas the other nitrogen atom of the ring, the N-3’ atom, is required to a lesser extent.
How is the dipolar carbanion of thiamine pyrophosphate formed?
Three tautomeric forms of the aminopyrimidine ring can be identified in the enzyme-bound coenzyme not involved in the reaction:
- the canonical 4’-aminopyrimidine tautomer;
- the N-1 protonated form, that is, 4-aminopyrimidinium ion;
- the 1’,4’-iminopyrimidine tautomer.
It seems that the 1′,4′-imino tautomer is the tautomer that undergoes deprotonation, before the entry of the substrate into the active site. The C-2 of the thiazolium ring is “much more acidic than most =C-H groups found in other molecules”. The higher acidity, i.e., the fact that the C-2 proton is easily dissociable, is due to the presence of the quaternary nitrogen on the thiazolium ring, a positively charged nitrogen atom able to electrostatically stabilize the resulting carbanion. In the deprotonation reaction, the amino group of the aminopyrimidine ring seems to play an essential role: it acts as a base and is suitably positioned to accept the proton. However, in the 4’-aminopyrimidine tautomer one of its protons sterically collides with the C-2 proton; in addition, its pK is too low to carry out the deprotonation efficiently.
A mechanism was therefore proposed, in which the side chain of a conserved glutamate residue, for example βGlu59 in B. stearothermophilus, or Glu51 in Saccharomyces uvarum (brewer’s yeast) pyruvate decarboxylase, donates a proton to the aminopyrimidine, converting it to its 1′,4′-iminopyrimidine tautomer that, accepting the C-2 proton, returns to the canonical 4′-aminopyrimidinic form and allows the formation of the carbanion.
Note: carbanion formation on C-2 is a consequence of an intramolecular proton transfer.
Deprotonation of thiamine pyrophosphate
The loss of the C-2 proton of the thiazolium ring leads, from a positively charged ring, to a dipolar ion, or zwitterion. This change in state of charge triggers a conformational change in one of the two conserved loops at the entrance of the active site channel, specifically, the inner of these loops, that, in turn, leads to the closure of the channel to the surrounding water environment. In this closed conformation the thiazolium carbanion is protected against electrophiles.
To sum up: the deprotonation of thiamine pyrophosphate leads to the closure of the active site and the protection of the newly formed dipolar carbanion, that is, TPP-dependent enzymes would be only active in closed conformation.
Conversely, in the other active site, thiamine pyrophosphate is not in the ylid form, the channel is open, and the site is inactive.
Reaction of dihydrolipoyl transacetylase
In the reaction sequence catalyzed by components of the pyruvate dehydrogenase complex, dihydrolipoyl transacetylase catalyzes the third step, namely, the transfer of the acetyl group from acetyl-dihydrolipoamide to CoA to form acetyl-CoA and dihydrolipoamide, the fully reduced form of lipoamide, the dithiol.
It should be noted that the acetyl group, initially bound by ester linkage to one of the –SH group of lipoamide is next bound to the –SH group of coenzyme A, again by ester bond, hence the term transesterification.
Catalytic mechanism of dihydrolipoyl transacetylase
During the reaction, the sulfhydryl group of coenzyme A carries out a nucleophilic attack on the carbonyl carbon of the acetyl group of acetyl dihydrolipoamide-dihydrolipoyl transacetylase to form a transient tetrahedral intermediate, that “decomposes” to dihydrolipoamide-dihydrolipoyl transacetylase and acetyl-CoA.
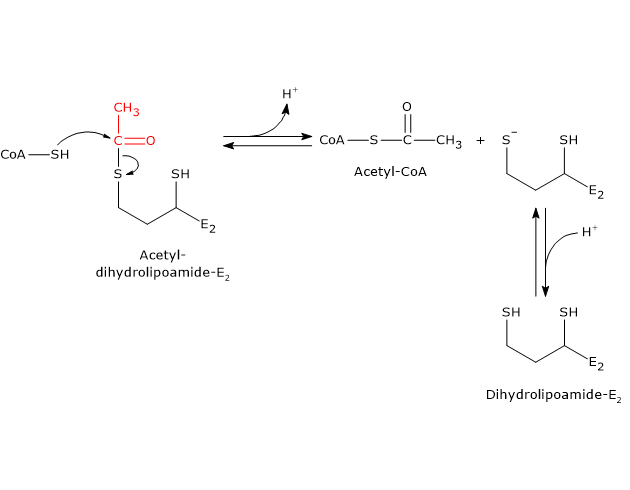
As previously said, the mobility of the lipoyllysyl arm plays a central role in the reaction mechanism.
Reaction of dihydrolipoyl dehydrogenase
In the reaction sequence catalyzed by components of the pyruvate dehydrogenase complex, dihydrolipoyl dehydrogenase catalyzes the fourth and fifth steps.
The enzyme catalyzes electron transfers needed to regenerate the disulfide bridge of the lipoyl group of dihydrolipoyl transacetylase, that is, to regenerate the oxidized form of the prosthetic group, and thus completing the catalytic cycle of the transacetylase.
The reaction has a ping-pong catalytic mechanism: it occurs in two successive half-reaction, in which each of the two substrates, NAD+ and dihydrolipoamide, reacts in the absence of the other. Moreover during the first half-reaction, the release of the first product and the formation of an enzyme intermediate complex occur before the second substrate binds, while the enzyme underogoes a structural change, whereas in the second half-reaction the release of the second product and the return of the enzyme to its starting state, again, via a structural change, occur.
Considering the ping-pong kinetic mechanism of dihydrolipoyl dehydrogenase:
- in the first half-reaction the oxidation of dihydrolipoamide to lipoamide occurs;
- in the second half-reaction the reduction of NAD+ to NADH occurs.
Catalytic mechanism of dihydrolipoyl dehydrogenase
Below, the reaction mechanism of P. putida dihydrolipoyl dehydrogenase is described.
In the first half-reaction, the oxidized dihydrolipoyl dehydrogenase (E), i.e., the enzyme with the disulfide bridge between Cys43 and Cys48, binds dihydrolipoamide (LH2) to form the enzyme-dihydrolipoamide complex (E●LH2). At this point, a sulfur atom of dihydrolipoamide carries out a nucleophilic attack on the sulfur of Cys43, to form the disulfide bridge lipoamide-Cys43 (E-S-S-L), while the sulfur of Cys48 is released as a thiolate ion (S–48).
The proton on the second thiol group of lipoamide is then abstracted by histidine (Hys) 451, that acts as a general acid-base catalyst, leading to the formation of a second thiolate ion, this time on the lipoamide (E-S-S-L ●S–), that, through a nucleophilic attack, displaces the sulfur of Cys43, S43, aided in this by general acid catalysis by Hys451 which donates a proton to S43. The catalytic action of Hys451 is essential, as demonstrated by mutagenesis studies in which its substitution with a glutamine residue causes the enzyme to retain ∼ 0.4% of the wild-type catalytic activity.
Then, thiolate anion S–48 contacts, through non-covalent interactions, the flavin ring near 4a position, i.e., an electron pair of S–48, which acts as electron donor, is partially transferred to the oxidized flavin ring, which, in turn, is the electron acceptor. The resulting structure is called charge-transfer complex.
Meanwhile, the phenolic side chain of the Tyr181 continues to hinder access to the flavin ring, thus protecting it from oxidation by O2.
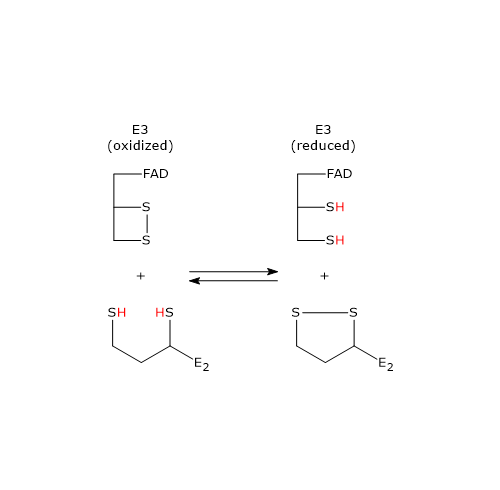
To sum up, what occurs is an interchange reaction of disulfide bridges leading to the formation of the oxidized form of lipoamide, the first product, which is released, and the reduced form of the dihydrolipoyl dehydrogenase.
The second half-reaction involves the reduction of NAD+ to NADH + H+ by electron transfer from the reactive disulfide of the enzyme via FAD.
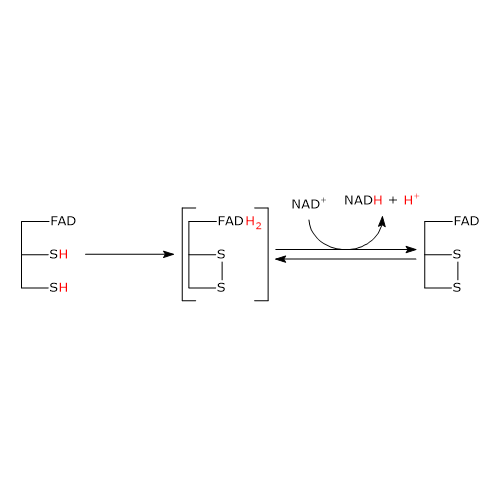
It begins with the entry of NAD+ into the active site and its binding to form the EH2●NAD+ complex. It should be noted that the entry of the coenzyme causes the phenolic side chain of the Tyr181 to be pushed aside by the nicotinamide ring.
Following the collapse of the charge-transfer complex, a covalent bond is formed between the flavin atom C-4a and S48, to which the extraction of a proton from S43 by the flavin atom N-5 is accompanied, with the formation of the corresponding thiolate anion, S–43.
S–43carries out a nucleophilic attack on S48, leading to the formation of the redox-active disulphide bridge between Cys43 and Cys48, followed by the breakdown of the covalent bond between S48 and the flavin atom C-4a to form reduced FADH anion, FADH–, with negative charge on atom N-1. It should be noted that dihydrolipoyl dehydrogenase is in the oxidized form (E).
FADH– has a transient existence because the proton bound to its N-5 is instantly transferred, as hydride ion, to the nicotinamide atom C-4, that is juxtaposed to flavin atom N-5. This leads to the formation of FAD and of the second product of the reaction, NADH, which is released.
To sum up, what occurs is that the electrons removed from the hydroxyethyl group, which derives from pyruvate, pass, via FAD, to NAD+. The catalytic cycle of dihydrolipoyl dehydrogenase is therefore completed, being the enzyme and its coenzymes in their oxidized form. At this point, the catalytic cycle of the entire pyruvate dehydrogenase complex is completed, too, and the complex is ready for a new reaction cycle.
Note: unlike the thiazolium ring of thiamine pyrophosphate, FAD does not acts as electron trap or electron sink, but rather as an electron conduit between the redox-active disulphide, in its reduced form, and NAD+.
Note: the catalytic mechanism of dihydrolipoyl dehydrogenase has been determined in analogy with that of glutathione reductase (EC 1.8.1.7), at 33% identical and whose structure is more extensively characterized. It should, however, be noted that although the two enzymes catalyze similar reactions, these usually occur in opposite direction:
- dihydrolipoyl dehydrogenase uses NAD+ to oxidize two –SH groups to a disulfide (–S–S–);
- glutathione reductase uses NADPH to reduce a –S–S– to two thiol groups.
Nevertheless, their active sites are closely superimposable.
Regulation of the pyruvate dehydrogenase complex
In mammals, the regulation of the activity of the pyruvate dehydrogenase complex is essential, both in the fed and fasted states. In fact, the multienzyme complex plays a central role in metabolism because, catalyzing the irreversible oxidative decarboxylation of pyruvate, represents the entry point of the carbon flux from all carbohydrate sources as well as from ∼50% of carbon skeletons of glucogenic amino acids, that, as a whole, correspond to ∼60 percent of the daily calorie intake, into:
- the citric acid cycle, and therefore to the full oxidation to CO2;
- the synthesis of lipids (fed state) and acetylcholine.
The importance of the regulation of the conversion of pyruvate into acetyl-CoA is also underlined by the fact that mammals, although able to produce glucose from pyruvate, cannot synthesize it from acetyl-CoA, because of the irreversibility of pyruvate dehydrogenase reaction and the absence of alternative pathways. Then, the inhibition of the activity of the complex allows to spare glucose and the amino acids that can be converted into pyruvate, such as alanine, when other fuels, for example acetyl-CoA from fatty acid oxidation, are available.
This explains why the activity of the complex is carefully regulated by:
- feedback inhibition;
- nucleotides;
- covalent modifications, namely, phosphorylation and dephosphorylation of specific target proteins.
Regulation by feedback inhibition and energy status of the cell
The activity of the dephosphorylated form of the pyruvate dehydrogenase complex is regulated by feedback inhibition.
Acetyl-CoA and NADH allosterically inhibit the enzymes that catalyze their formation, dihydrolipoyl transacetylase and dihydrolipoyl dehydrogenase, respectively.
In addition, CoA and acetyl-CoA, as well as NAD+ and NADH, compete for binding sites on E2 and E3, respectively, that catalyze reversible reactions. This means that, in the presence of high ratios of [Acetyl-CoA]/[CoA] and [NADH]/ [NAD+], the reactions of transacetylation and dehydrogenation work in reverse; therefore, dihydrolipoyl transacetylase cannot accept the hydroxyethyl group from TPP because it is maintained in the acetylated form. This cause thiamine pyrophosphate to remain bound to pyruvate dehydrogenase in its hydroxyethyl form, which, in turn, decreases the rate of pyruvate decarboxylation. Hence, high ratios of [Acetyl-CoA]/[CoA] and [NADH]/[NAD+] indirectly influence pyruvate dehydrogenase activity.
Acetyl-CoA and NADH are also produced by fatty acid oxidation, which takes place, like the reactions of the pyruvate dehydrogenase complex, within the mitochondrion. This means that the cell, by regulating the activity of the multienzyme complex, preserves carbohydrate stores when fatty acids are available for energy. For example, during the fasted state, liver, skeletal muscle and many other organs and tissues rely primarily on fatty acid oxidation for energy. Conversely, the activity of the multienzyme complex is increased in the fed state, when many different types of cells and tissues mainly use glucose as a fuel.
More generally, when the production of NADH and/or acetil-CoA exceeds the capacity of the cell to use them for ATP production, the activity of the pyruvate dehydrogenase complex is inhibited. The same is true when there is no need for additional ATP to be produced. Infact, the activity of multienzyme complex is also sensitive to the energy charge of the cell. Through allosteric mechanisms, high ATP levels inhibit the activity of the pyruvate dehydrogenase component of the complex, whereas high ADP levels, that signs that the energy charge of the cell may become low, activate it, thus committing the carbon skeleton of carbohydrates and some amino acids to energy production.
Note: in the skeletal muscle, the activity of the pyruvate dehydrogenase complex increases with increased aerobic activity, resulting in a in greater dependence on glucose as a fuel source.
Regulation by phosphorylation/dephosphorylation
Unlike prokaryotes, in mammals the activity of the pyruvate dehydrogenase complex is also regulated by covalent modifications, i.e., phosphorylation and dephosphorylation of three specific serine residues of the α subunit of pyruvate dehydrogenase, the enzyme that catalyzes the first, irreversible step of the overall reaction sequence.
Note: as mammalian pyruvate dehydrogenase is an heterotetramer, there are six potential phosphorylation sites.
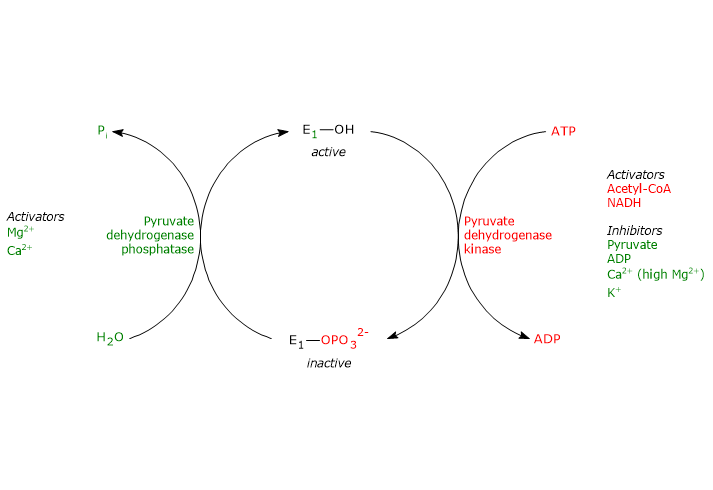
Phosphorylation, which inactivates pyruvate dehydrogenase, and then blocks the overall reaction sequence, is catalyzed by pyruvate dehydrogenase kinase. Two of the aforementioned serine residues are located on the more C-terminal loop, at the entrance of the substrate channel leading to the respective active site, and the phosphorylation of only one of them inactivates the pyruvate dehydrogenase, hence demonstrating the out of phase coupling between its active sites.
Conversely, in the dephosphorylated state the complex is active. Dephosphorylation is catalyzed by a specific protein phosphatase, the pyruvate dehydrogenase phosphatase.
The activities of pyruvate dehydrogenase kinase and pyruvate dehydrogenase phosphatase are in turn subject to allosteric regulation by several modulators.
Regulation of pyruvate dehydrogenase kinase
The activity of pyruvate dehydrogenase kinase depends on the ratios of [NADH]/[NAD+], [acetyl-CoA]/[CoA], and [ATP]/[ADP] , as well as on the pyruvate concentration, in the mitochondrial matrix.
- High ratios of [NADH]/[NAD+] and [acetyl-CoA]/[CoA], as during the oxidation of fatty acids and ketone bodies, activate the kinase, pyruvate dehydrogenase is phosphorylated, and the pyruvate dehydrogenase complex is inhibited. This allows tissues, such as cardiac muscle, to preserve glucose when fatty acids and/or ketone bodies are utilized for energy, because acetyl-CoA synthesis from pyruvate, and hence from carbohydrates (and some amino acids) is turned off.
Conversely, when the concentrations of NAD+ and coenzyme A are high the activity of the kinase is inhibited and the multienzyme complex is active.
Therefore, acetyl-CoA and NADH, two of the three end products of the reactions catalyzed by the pyruvate dehydrogenase complex, allosterically control their synthesis by regulating directly and indirectly, by regulating the activity of pyruvate dehydrogenase kinase, the activity of the complex. - A high ratio of [ATP]/[ADP] activates the kinase, and then inhibits the pyruvate dehydrogenase complex.
Note: unlike many other kinases, such as those involved in the control of glycogen metabolism, pyruvate dehydrogenase kinase is not regulated by cAMP levels, but by molecules that signal changes in energy status of the cell and in the availability of biosynthetic intermediates: ATP and NADH, and acetyl-CoA, respectively. - Pyruvate allosterically inhibits pyruvate dehydrogenase kinase.
When its levels are high, it binds to kinase and inactivates it, pyruvate dehydrogenase is not phosphorylated, and the pyruvate dehydrogenase complex remains active. - Pyruvate dehydrogenase kinase is also activated by interaction with dihydrolipoyl transacetylase in its acetylated form, i.e. when acetyl-dihydrolipoamide is present.
Other activators of the kinase is potassium and magnesium ions.
Regulation of pyruvate dehydrogenase phosphatase
The activity of pyruvate dehydrogenase phosphatase depends on the ratios of [NADH]/[NAD+] and [acetyl-CoA]/[CoA], as well as on [Ca2+], in the mitochondrial matrix.
- Low ratios of [NADH]/[NAD+] and [acetyl-CoA]/[CoA] activate the phosphatase, pyruvate dehydrogenase is dephosphorylated, and the pyruvate dehydrogenase complex is activated.
Conversely, when the aforesaid ratios are high, phosphatase activity is reduced, kinase activity is increased, and the multienzyme complex is inhibited. - Calcium ion activates pyruvate dehydrogenase phosphatase.
Ca2+ is an important second messenger that signals the cell requires more energy. Therefore, when its levels are high, as in cardiac muscle cells after epinephrine stimulation or in skeletal muscle cells during the muscular contraction, the phosphatase is active, the complex is dephosphorylated, and then active. - Insulin, too, is involved in the control of the activity of the pyruvate dehydrogenase complex through the activation of pyruvate dehydrogenase phosphatase. The hormone, in response to increases in blood glucose, stimulates glycogen synthesis and the synthesis of acetyl-CoA, a precursor in the synthesis of lipids.
Fasting and subsequent refeeding, too, affect the activity of the multienzyme complex.
In tissues such as skeletal muscle, cardiac muscle or kidney, fasting significantly decreases the activity of the complex, whereas refeeding reverses the inhibition of fasting.
In the brain, however, these variations are not observed because the activity of pyruvate dehydrogenase complex is essential for ATP production.
References
- Frank R.A.W., Titman C.M., Pratap J.V., Luisi B.F., and Perham R.N. A molecular switch and proton wire synchronize the active sites in thiamine enzymes. Science 2004;306(5697):872-876. doi:10.1126/science.1101030
- Garrett R.H., Grisham C.M. Biochemistry. 4th Edition. Brooks/Cole, Cengage Learning, 2010
- Gray L.R., Tompkins S.C., Taylor E.R. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-2604. doi:10.1007/s00018-013-1539-2
- McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
- Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 6th Edition. W.H. Freeman and Company, 2012
- Nemeria N.S., Chakraborty S., Balakrishnan A., and Frank Jordan. Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at individual steps. FEBS J 2009;276:2432-2446. doi:10.1111/j.1742-4658.2009.06964.x
- Patel M.S. and Korotchkina L.G. Regulation of the pyruvate dehydrogenase complex. Biochem Soc T 2006;34(2):217-222. doi:10.1042/bst0340217
- Patel M.S., Nemeria N.S., Furey W., and Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem 2014;289(24):16615-16623. doi:10.1074/jbc.R114.563148
- Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009
- Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 3rd Edition. Elsevier health sciences, 2012
- Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011
- Wang J., Nemeria N.S., Chandrasekhar K., Kumaran S., Arjunan P., Reynolds S., Calero G., Brukh R., Kakalis L., Furey W., and Jordan F. Structure and function of the catalytic domain of the dihydrolipoyl acetyltransferase component in Escherichia coli pyruvate dehydrogenase complex. J Biol Chem 2014;289(22):15215-15230. doi:10.1074/jbc.M113.544080
- Zhou Z.H., McCarthy D.B., O’Connor C.M., Reed L.J., and J.K. Stoops. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA 2001;98(26):14802-14807. doi:10.1073/pnas.011597698