Gluconeogenesis is a metabolic pathway that leads to the synthesis of glucose (Glu) from pyruvate (Pyr), the conjugate base of pyruvic acid, and other non-carbohydrate precursors, even in non-photosynthetic organisms.[1]
It occurs in all microorganisms, fungi, plants, and animals, and the reactions are essentially the same, leading to the synthesis of one glucose molecule from two pyruvate molecules. It is essentially glycolysis in reverse, sharing seven enzymes with it, but proceeding from pyruvate to glucose.[2]
Glycogenolysis is clearly distinct from gluconeogenesis, as it does not result in the de novo synthesis of glucose from non-carbohydrate precursors, as illustrated by its overall reaction:[3]
Glycogen or (glucose)n → n glucose 1-phosphate molecules.
The following discussion will focus on gluconeogenesis in higher animals, particularly in the mammalian liver.
Summary: Key Points
- Definition: the metabolic pathway responsible for synthesizing glucose from non-carbohydrate precursors such as pyruvate, lactate, glycerol, and glucogenic amino acids.
- Biological function: essential for maintaining stable blood glucose levels, particularly to supply glucose to the brain and red blood cells during prolonged fasting.
- Mechanism: not a simple reversal of glycolysis; instead, it bypasses three irreversible glycolytic steps through the action of specific enzymes, including pyruvate carboxylase, phosphoenolpyruvate carboxykinase, and fructose-1,6-bisphosphatase.
- Main sites: occurs primarily in the liver (≈90%) and, to a lesser extent, in the renal cortex, involving the mitochondria, cytosol, and endoplasmic reticulum.
- Hormonal control: regulated mainly by the insulin-to-glucagon ratio and by allosteric effectors such as fructose-2,6-bisphosphate.
Contents
- Importance of gluconeogenesis
- Sites of gluconeogenesis
- Irreversible steps in the pathway
- From pyruvate to phosphoenolpyruvate
- From fructose 1,6-bisphosphate to fructose 6-phosphate
- From glucose 6-phosphate to glucose
- Energy cost of gluconeogenesis
- Coordinated regulation of gluconeogenesis and glycolysis
- Regulation of gluconeogenesis
- Precursors
- References
Importance of gluconeogenesis
Gluconeogenesis is essential for two main reasons: it maintains appropriate blood glucose levels when glycogen stores are low and no carbohydrates are available, and it preserves metabolic intermediates, such as pyruvate, needed for energy production.[4]
Maintaining blood glucose between 3.3 and 5.5 mmol/L (60–99 mg/dL) is critical, as many cells rely on glucose to meet their ATP needs. These include red blood cells, neurons, the renal medulla, skeletal muscle under low oxygen, the testes, the lens and cornea, and embryonic tissues.[5]
For example, the brain alone requires about 120 g/day of glucose, which represents:
- over 50% of the total body glucose stores (≈210 g), of which about 190 g are stored as liver and muscle glycogen, and 20 g are found free in body fluids;
- about 75% of the daily glucose requirement (≈160 g).[4]
During fasting, such as between meals or overnight, blood glucose levels are maintained within the normal range primarily through hepatic glycogenolysis, as well as the release of fatty acids from adipose tissue and ketone body production by the liver.
Fatty acids and ketone bodies are preferentially used by skeletal muscle, thereby sparing glucose for cells that depend entirely on it, especially red blood cells and neurons.
However, after approximately 18 hours of fasting, or during intense and prolonged exercise, glycogen stores become insufficient. At this point, in the absence of carbohydrate intake, gluconeogenesis becomes critical.[6]
Its importance is further highlighted by the fact that blood glucose levels below 2 mmol/L can lead to unconsciousness.[7]
Additionally, without gluconeogenesis, pyruvate would be lost, reducing the body’s capacity to produce ATP via aerobic respiration (more than 10 ATP molecules per pyruvate oxidized), making its conservation essential during energy stress.[1]
Sites of gluconeogenesis
In higher animals, gluconeogenesis occurs in the liver, kidney cortex, and enterocytes.[4]
Quantitatively, the liver is the major site, producing about 90% of synthesized glucose, followed by the kidney cortex with around 10%. The liver’s key role is largely due to its size; in fact, on a wet weight basis, the kidney cortex produces more glucose than the liver.[8]
In the kidney, gluconeogenesis takes place in the proximal tubule cells. Much of the glucose produced is consumed by the renal medulla, while the kidney’s role in maintaining blood glucose becomes more prominent during prolonged fasting or liver failure. Unlike the liver, the kidney lacks significant glycogen stores and contributes to glucose homeostasis only through gluconeogenesis, not glycogenolysis.
Only limited portions of the gluconeogenic pathway are active in skeletal muscle, cardiac muscle, and the brain, and at very low rates.
However, these tissues cannot release free glucose into the bloodstream because they lack glucose 6-phosphatase (EC 3.1.3.9), the enzyme responsible for the final step of gluconeogenesis. As a result, glucose 6-phosphate (G6P) produced, either via gluconeogenesis or glycogenolysis, remains trapped in the cell and is used only for internal energy needs or glycogen resynthesis. In the brain, this occurs mainly in astrocytes. The only direct contribution of these tissues to blood glucose maintenance, particularly skeletal muscle, due to its large mass (approximately 18 times that of the liver), comes from the limited release of free glucose via the debranching enzyme (EC 3.2.1.33) during glycogenolysis.[4]
Most gluconeogenic reactions occur in the cytosol, though some steps take place in the mitochondria, and the final step, catalyzed by glucose 6-phosphatase, occurs within the endoplasmic reticulum cisternae.[3]
Irreversible steps in the pathway
As previously mentioned, gluconeogenesis is essentially glycolysis in reverse. Of the ten reactions that constitute gluconeogenesis, seven are shared with glycolysis. These reactions have a ΔG close to zero and are therefore easily reversible. However, under intracellular conditions, the overall ΔG of glycolysis is approximately –63 kJ/mol (–15 kcal/mol), while that of gluconeogenesis is about –16 kJ/mol (–3.83 kcal/mol). This indicates that both pathways are irreversible in vivo.
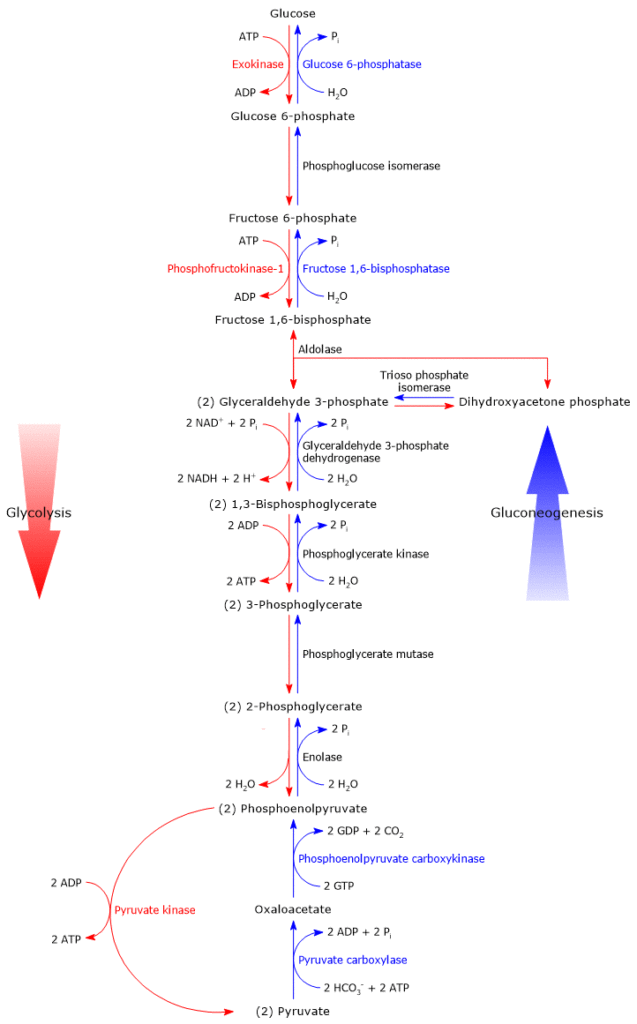
The irreversibility of glycolysis arises from three strongly exergonic reactions that cannot be used in gluconeogenesis.
- Phosphorylation of glucose to glucose 6-phosphate, catalyzed by hexokinase (EC 2.7.1.1) or glucokinase (EC 2.7.1.2).
- Phosphorylation of fructose 6-phosphate to fructose 1,6-bisphosphate (F1,6BP), catalyzed by phosphofructokinase-1 (PFK-1; EC 2.7.1.11).
- Conversion of phosphoenolpyruvate (PEP) to pyruvate, catalyzed by pyruvate kinase (EC 2.7.1.40).
| Reaction | Catalyzing enzyme | ΔG (kJ/mol) (kcal/mol) | ΔG°’ (kJ/mol) (kcal/mol) |
|---|---|---|---|
| Glu → G6P | Hexokinase or Glucokinase | –33.4 (–8.0) | –16.7 (–4.0) |
| F6P → F1,6BP | PFK-1 | –22.2 (–5.3) | –14.2 (–3.4) |
| PEP → Pyr | Pyruvate kinase | –16.7 (–4.0) | –31.4 (–7.5) |
In gluconeogenesis, these three steps are bypassed by enzymes that catalyze irreversible reactions in the direction of glucose synthesis.
These bypass reactions ensure that gluconeogenesis, like glycolysis, remains a thermodynamically irreversible pathway.[2]
The next sections will analyze these reactions in detail.
From pyruvate to phosphoenolpyruvate
The first step of gluconeogenesis that bypasses an irreversible step of glycolysis, namely the reaction catalyzed by pyruvate kinase, is the conversion of pyruvate to phosphoenolpyruvate.
This occurs through a two-step process catalyzed by:
- pyruvate carboxylase (EC 6.4.1.1)
- phosphoenolpyruvate carboxykinase (PEPCK; EC 4.1.1.32)
The overall reaction can be summarized as:[9]
Pyr → Oxaloacetate → PEP
Pyruvate carboxylase and oxaloacetate formation
Pyruvate carboxylase catalyzes the carboxylation of pyruvate to oxaloacetate, consuming one molecule of ATP in the process. The enzyme requires magnesium or manganese ions.
Pyr + HCO3–+ ATP → Oxaloacetate + ADP + Pi
Discovered in 1960 by Merton Utter, pyruvate carboxylase is a mitochondrial enzyme composed of four identical subunits, each containing a biotin prosthetic group. The biotin is covalently attached by an amide bond to the ε-amino group of a lysine residue and serves as a carrier of activated CO2 during the reaction. Each subunit also contains an allosteric site for acetyl-CoA, an essential activator of the enzyme.
In addition to its role in gluconeogenesis, this reaction also provides intermediates for the citric acid cycle.[10][11]
Phosphoenolpyruvate carboxykinase and PEP formation
Phosphoenolpyruvate carboxykinase catalyzes the decarboxylation and phosphorylation of oxaloacetate to form PEP. This reaction requires GTP as a phosphate donor and the presence of magnesium and manganese ions.[12]
Oxaloacetate + GTP → PEP + CO2 + GDP
The CO2 removed in this step is the same one added to pyruvate by pyruvate carboxylase. This carboxylation–decarboxylation sequence is crucial, as it facilitates the formation of PEP by making the reaction energetically favorable.[1]
More generally, such sequences are used to drive otherwise endergonic reactions and are also found in other metabolic pathways like the citric acid cycle, the pentose phosphate pathway, and fatty acid synthesis.[2]
Energetics of the overall conversion
The net reaction catalyzed by pyruvate carboxylase and PEPCK is:
Pyr + ATP + GTP + HCO3– → PEP + ADP + GDP + Pi + CO2
The ΔG°’ of this combined reaction is +0.9 kJ/mol (0.2 kcal/mol).
In contrast, the ΔG°’ for the reverse glycolytic reaction catalyzed by pyruvate kinase is +31.4 kJ/mol (7.5 kcal/mol).
Under intracellular conditions, however, the actual ΔG is highly negative (–25 kJ/mol, or –6 kcal/mol), due to rapid consumption of PEP, which keeps its concentration low.
Thus, the formation of PEP from pyruvate is effectively irreversible in vivo.[1]
Phosphoenolpyruvate precursors: pyruvate or alanine
Gluconeogenesis can proceed from various glucogenic precursors. This section examines the specific steps involved when pyruvate or alanine serve as the starting materials, highlighting how the pathway adapts according to the metabolic conditions.
Mitochondrial transport and conversion of pyruvate and alanine
The reactions described below predominate when pyruvate or alanine is the glucogenic precursor.
Since pyruvate carboxylase is a mitochondrial enzyme, pyruvate must first be transported from the cytosol into the mitochondrial matrix. This transport is mediated by specific proteins located in the inner mitochondrial membrane, namely MPC1 and MPC2. These two proteins associate to form a hetero-oligomeric complex that facilitates pyruvate import into mitochondria.[13]
Alternatively, pyruvate can be generated inside the mitochondrial matrix via transamination of alanine, catalyzed by alanine aminotransferase (EC 2.6.1.2). In this reaction, the amino group of alanine is transferred to α-ketoglutarate, producing glutamate and pyruvate. The resulting amino group is subsequently excreted as urea through the urea cycle.[14]
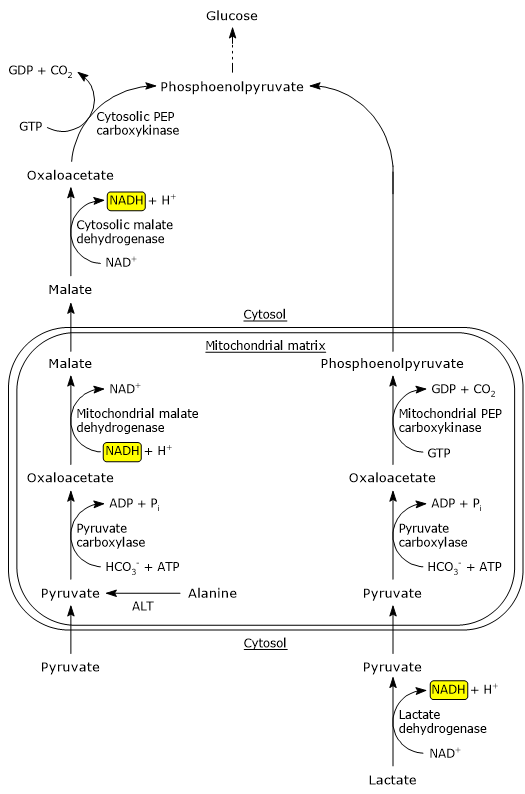
Oxaloacetate transport and conversion to PEP
Since most gluconeogenic enzymes (with the exception of glucose-6-phosphatase) are cytosolic, the oxaloacetate produced in the mitochondrial matrix must be transferred to the cytosol. However, oxaloacetate itself cannot cross the inner mitochondrial membrane directly.
To overcome this, it is reduced to malate in a reaction catalyzed by mitochondrial malate dehydrogenase (EC 1.1.1.37), an enzyme also involved in the citric acid cycle. This reaction uses NADH as a reducing agent, which is oxidized to NAD+.
Oxaloacetate + NADH + H+ ⇄ Malate + NAD+
Although the ΔG°’ of this reaction is strongly positive, under physiological conditions the ΔG is close to zero, making the reaction reversible and efficient in vivo.[2]
Malate is then transported into the cytosol via the malate-α-ketoglutarate transporter, a component of the malate-aspartate shuttle. Once in the cytosol, malate is re-oxidized to oxaloacetate by cytosolic malate dehydrogenase, regenerating NADH.
Malate + NAD+ → Oxaloacetate + NADH + H+
Note: the malate-aspartate shuttle is the most active mechanism for transferring reducing equivalents (NADH) from the cytosol into mitochondria. It operates in tissues such as the liver, kidney, and heart.[3]
The reverse use of this shuttle in gluconeogenesis allows NADH to be generated in the cytosol, which is essential for the reaction catalyzed by glyceraldehyde 3-phosphate dehydrogenase (EC 1.2.1.12). In fact, cytosolic NADH is normally present at very low concentrations, with a [NADH]/[NAD+] ratio of about 8 × 10−4, roughly 100,000 times lower than in mitochondria.[15]
Finally, the cytosolic oxaloacetate is converted to phosphoenolpyruvate by PEP carboxykinase.
Phosphoenolpyruvate precursor: lactate
Lactate is one of the major gluconeogenic precursors. It is produced, for example, by:
- red blood cells, which rely entirely on anaerobic glycolysis for ATP production;
- skeletal muscle during intense exercise, that is, under low-oxygen conditions when the rate of glycolysis exceeds that of the citric acid cycle and oxidative phosphorylation.[9]
When lactate serves as the gluconeogenic precursor, PEP synthesis proceeds through a different pathway than the one described for pyruvate or alanine.
In the hepatocyte cytosol, where the NAD+ concentration is high, lactate is oxidized to pyruvate by the liver isoenzyme of lactate dehydrogenase (EC 1.1.1.27). During this reaction, NAD+ is reduced to NADH.
Lactate + NAD+ → Pyr + NADH + H+
The generation of cytosolic NADH makes the export of reducing equivalents from mitochondria unnecessary.
Pyruvate then enters the mitochondrial matrix, where it is converted to oxaloacetate by pyruvate carboxylase. In this case, oxaloacetate is directly converted to PEP by the mitochondrial isoform of PEP carboxykinase. PEP is then transported out of the mitochondria via an anion transporter located in the inner mitochondrial membrane and continues along the gluconeogenic pathway in the cytosol.[3]
Note: the synthesis of glucose from lactate represents the hepatic phase of the Cori cycle, which links peripheral tissues to the liver in the recycling of lactate.[16]
From fructose 1,6-bisphosphate to fructose 6-phosphate
The second step of gluconeogenesis that bypasses an irreversible reaction of the glycolytic pathway, specifically the one catalyzed by phosphofructokinase-1, is the dephosphorylation of fructose 1,6-bisphosphate to fructose 6-phosphate.[5]
This reaction is catalyzed by fructose 1,6-bisphosphatase (FBPase-1; EC 3.1.3.11), a Mg2+-dependent enzyme located in the cytosol. It involves the hydrolysis of the phosphate group at the C-1 position of fructose 1,6-bisphosphate and does not generate ATP.
F1,6BP + H2O → F6P + Pi
The ΔG°’ for the reaction is –16.3 kJ/mol (–3.9 kcal/mol), indicating that it is irreversible under cellular conditions.[2]
From glucose 6-phosphate to glucose
The third step of gluconeogenesis that bypasses an irreversible reaction of the glycolytic pathway, specifically the one catalyzed by hexokinase or glucokinase, is the dephosphorylation of glucose 6-phosphate to glucose.[5]
This reaction is catalyzed by the catalytic subunit of glucose 6-phosphatase, part of a protein complex located in the endoplasmic reticulum membrane of hepatocytes, enterocytes, and the proximal tubule cells of the kidney.[8] The glucose 6-phosphatase complex consists of the catalytic subunit and a glucose 6-phosphate transporter known as T1 (glucose 6-phosphate translocase).
Importantly, the enzyme’s active site faces the lumen of the endoplasmic reticulum. This means that glucose is released inside the endoplasmic reticulum, not in the cytosol.[17]
Mechanism and regulation of glucose release
Glucose 6-phosphate, derived either from gluconeogenesis via the action of glucose 6-phosphate isomerase (EC 5.3.1.9), or from glycogenolysis via phosphoglucomutase (EC 5.4.2.2), is found in the cytosol and must be transported into the endoplasmic reticulum lumen to be dephosphorylated. This transport is mediated by T1 translocase.
Once inside the endoplasmic reticulum, the Mg2+-dependent catalytic subunit catalyzes the hydrolysis of the phosphate ester bond.[17]
G6P + H2O → Glu + Pi
As with the reaction catalyzed by FBPase-1, this is an irreversible reaction. The ΔG°′ is –13.8 kJ/mol (–3.3 kcal/mol). In contrast, the reverse reaction, catalyzed by hexokinase or glucokinase, would require the transfer of a phosphate group from glucose 6-phosphate to ADP, which would be highly endergonic (ΔG = +33.4 kJ/mol, or +8 kcal/mol).[2]
To prevent interference with glycolysis, which occurs in the cytosol, the glucose 6-phosphatase activity is spatially restricted to the endoplasmic reticulum lumen, ensuring compartmental separation of opposing pathways.[4]
Glucose and inorganic phosphate are exported from the endoplasmic reticulum to the cytosol by distinct transporters, likely T2, for glucose, and T3, an anion transporter for inorganic phosphate. Finally, glucose leaves the hepatocyte via the GLUT2 transporter, enters the bloodstream, and is delivered to tissues in need.[17]
Energy cost of gluconeogenesis
Like glycolysis, gluconeogenesis involves several energetically costly steps, particularly those that bypass the irreversible reactions of glycolysis.
In total, six high-energy phosphate bonds are consumed in the synthesis of one molecule of glucose from two molecules of pyruvate: four ATP and two GTP.
In addition, two molecules of NADH are required for the reduction of 1,3-bisphosphoglycerate to glyceraldehyde 3-phosphate, in the reaction catalyzed by glyceraldehyde 3-phosphate dehydrogenase.
The oxidation of NADH used in gluconeogenesis implies a loss of potential ATP that would otherwise be produced via oxidative phosphorylation, approximately five molecules of ATP are not synthesized due to the diversion of NADH from the electron transport chain.
These energetic demands reinforce the concept that gluconeogenesis is not simply glycolysis in reverse. If it were, it would require only the energy equivalent of two ATP molecules, as shown in the overall glycolytic equation.
Glu + 2 ADP + 2 Pi + 2 NAD+ → 2 Pyr + 2 ATP + 2 NADH + 2 H+ + 2 H2O
In contrast, the overall reaction of gluconeogenesis is:[2][3]
2 Pyr + 4 ATP + 2 GTP + 2 NADH + 2 H+ + 4 H2O → Glucose + 4 ADP + 2 GDP + 6 Pi + 2 NAD+
In the liver, this energy is primarily provided by the oxidation of fatty acids. Under certain metabolic conditions, especially during prolonged fasting or high-protein intake, the oxidation of amino acid carbon skeletons can also contribute significantly to ATP and GTP production. The predominant source of energy depends on the available metabolic fuels at the time.[6]
Coordinated regulation of gluconeogenesis and glycolysis
If glycolysis and gluconeogenesis were simultaneously active at high rates within the same cell, the net result would be the consumption of ATP and the generation of heat, particularly at the irreversible steps of both pathways, with no productive metabolic outcome.
For example, consider the opposing reactions catalyzed by phosphofructokinase-1 and fructose 1,6-bisphosphatase-1.
PFK-1 reaction:
ATP + F6P → ADP + F1,6BP
FBPase-1 reaction:
F1,6BP + H2O → F6P + Pi
Sum of the two reactions:[1]
ATP + H2O → ADP + Pi + Heat
These two reactions, if occurring simultaneously in opposite directions, result in a futile cycle (also called a substrate cycle), leading to a wasteful dissipation of energy.[18]
To avoid this, the cell tightly regulates these pathways through three main mechanisms:
- allosteric regulation
- covalent modification (e.g., phosphorylation and dephosphorylation);
- changes in enzyme concentration, regulated at the transcriptional or degradative level.
Allosteric regulation is rapid and reversible, occurring within milliseconds. In contrast, covalent modifications are typically triggered by extracellular signals, such as hormones like insulin, glucagon, or epinephrine, and occur over seconds to minutes. Regulation via enzyme synthesis or degradation takes even longer, on the order of hours.
This multi-layered regulation ensures that when pyruvate is directed into gluconeogenesis, glycolytic flux is simultaneously reduced, and vice versa, allowing for metabolic coordination in response to the cell’s energy status and systemic hormonal cues.[1]
Regulation of gluconeogenesis
The regulation of gluconeogenesis, like that of glycolysis, primarily targets the enzymes that are unique to each pathway, rather than those that are shared between them.
In glycolysis, the major regulatory control points are the reactions catalyzed by PFK-1 and pyruvate kinase. In contrast, gluconeogenesis is mainly regulated at the steps catalyzed by fructose 1,6-bisphosphatase and pyruvate carboxylase.
The other two enzymes unique to gluconeogenesis, glucose 6-phosphatase and phosphoenolpyruvate carboxykinase, are regulated primarily at the transcriptional level, in response to hormonal and metabolic signals.[4]
Pyruvate carboxylase
In mitochondria, pyruvate can be directed toward two major metabolic fates:
- conversion to acetyl-CoA via the pyruvate dehydrogenase complex (PDC) linking glycolysis to the citric acid cycle;[19]
- carboxylation to oxaloacetate via pyruvate carboxylase, initiating the gluconeogenic pathway.
The fate of pyruvate depends largely on the availability of acetyl-CoA, which in turn reflects the supply of fatty acids in the mitochondrion.
When fatty acids are abundant, their β-oxidation generates acetyl-CoA, which enters the citric acid cycle and produces GTP and NADH. If the cell’s energy needs are met, oxidative phosphorylation slows down, leading to an increase in the [NADH]/[NAD+] ratio. This elevated ratio inhibits the citric acid cycle, causing acetyl-CoA to accumulate in the mitochondrial matrix.
Acetyl-CoA acts as a key metabolic signal:
- it is a positive allosteric effector of pyruvate carboxylase, promoting gluconeogenesis;
- it is a negative allosteric effector of pyruvate kinase, inhibiting glycolysis;
- it also inhibits the pyruvate dehydrogenase complex through feedback inhibition and via phosphorylation, activating a specific PDC kinase.
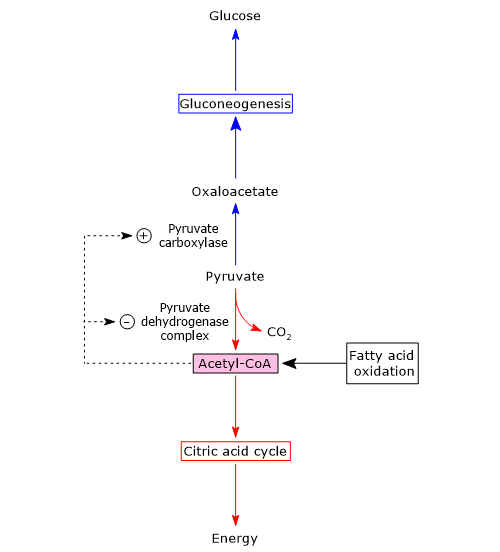
Thus, when the cellular energy charge is high, the conversion of pyruvate to acetyl-CoA slows down, while its conversion to oxaloacetate, and ultimately glucose, is promoted. In this context, acetyl-CoA acts as a metabolic signal indicating that further glucose oxidation is unnecessary, and that glucogenic precursors should be directed toward glucose synthesis and storage.
Conversely, when acetyl-CoA levels are low, pyruvate is preferentially directed toward energy production:
- pyruvate kinase and the pyruvate dehydrogenase complex are more active;
- the flow of carbon into the citric acid cycle increases;
- cellular energy needs are met through oxidative metabolism.[10][20]
In summary, pyruvate carboxylase represents the first major control point of gluconeogenesis, determining whether pyruvate is used for energy production or diverted toward glucose synthesis, based on the energetic status of the cell.[21]
Fructose 1,6-bisphosphatase
The second major control point in gluconeogenesis is the reaction catalyzed by fructose 1,6-bisphosphatase. This enzyme is allosterically inhibited by AMP, meaning that when AMP levels are high, and consequently ATP levels are low, gluconeogenesis slows down. Thus, as previously mentioned, FBPase-1 is active only when the cellular energy charge is sufficiently high to support de novo glucose synthesis.[22]
In contrast, phosphofructokinase-1, the glycolytic counterpart, is allosterically activated by AMP and ADP, and inhibited by ATP and citrate, the latter being a product of acetyl-CoA and oxaloacetate condensation.[23]
Therefore:
- when AMP levels are high, glycolysis is favored, and gluconeogenesis is inhibited;
- when ATP, acetyl-CoA, or citrate levels are high, gluconeogenesis is promoted, and glycolysis slows down.
The accumulation of citrate, in particular, signals a slowdown in the citric acid cycle and facilitates the diversion of pyruvate toward glucose synthesis.[21][24]
PFK-1, FBPase-1, and fructose 2,6-bisphosphate
The liver plays a central role in maintaining blood glucose homeostasis, requiring tight coordination between glucose consumption and production. Two hormones are primarily responsible for this regulation: insulin and glucagon.[21]
Their intracellular effects are largely mediated through fructose 2,6-bisphosphate (F2,6BP), a potent allosteric activator of PFK-1 and inhibitor of FBPase-1. F2,6BP is structurally similar to fructose 1,6-bisphosphate but is not an intermediate in either glycolysis or gluconeogenesis.[1]
Discovered in 1980 by Emile Van Schaftingen and Henri-Gery Hers, F2,6BP was first identified as a powerful activator of PFK-1. The following year, it was also shown to be a strong inhibitor of FBPase-1.
F2,6BP binds to an allosteric site on PFK-1, reducing its affinity for ATP and citrate (inhibitors) while increasing its affinity for fructose 6-phosphate (its substrate). Without F2,6BP, and under physiological conditions with ATP and citrate present, PFK-1 is nearly inactive. The presence of F2,6BP activates PFK-1 and stimulates glycolysis, while simultaneously inhibiting FBPase-1, even in the absence of AMP. The effects of AMP and F2,6BP on FBPase-1 are synergistic.[25][26]
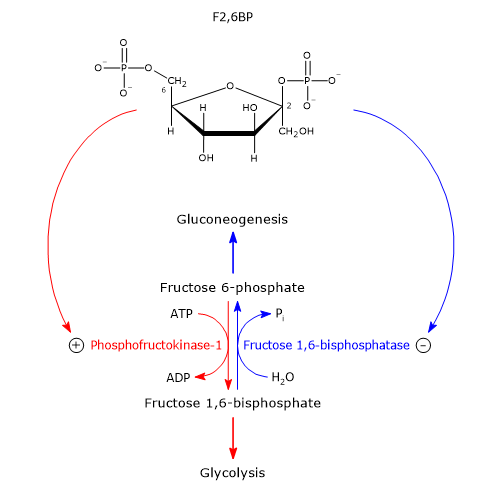
Regulation of F2,6BP levels
The cellular concentration of F2,6BP is regulated by the balance between its synthesis and degradation.
- Synthesis: catalyzed by phosphofructokinase-2 (PFK-2), from fructose 6-phosphate (EC 2.7.1.105).
- Degradation: catalyzed by fructose 2,6-bisphosphatase (FBPase-2), converting F2,6BP back to fructose 6-phosphate (EC 3.1.3.46).
These two enzymatic activities reside on a single bifunctional protein, often called the PFK-2/FBPase-2 tandem enzyme. In the liver, the balance of these two functions is hormonally regulated.[27]
Glucagon
After binding to its specific membrane receptors, glucagon stimulates adenylate cyclase (EC 4.6.1.1) to synthesize 3′,5′-cyclic adenosine monophosphate (cAMP). This second messenger activates cAMP-dependent protein kinase, also known as protein kinase A (PKA) (EC 2.7.11.11).
PKA catalyzes the phosphorylation of a specific serine residue (Ser32) on the bifunctional enzyme PFK-2/FBPase-2, using one molecule of ATP. Phosphorylation increases the enzyme’s fructose 2,6-bisphosphatase activity while decreasing its phosphofructokinase-2 activity. This shift is partly due to an increase in the Km for fructose 6-phosphate, which reduces the enzyme’s affinity for its substrate.
As a result, the intracellular concentration of fructose 2,6-bisphosphate decreases. Since this molecule is a potent activator of PFK-1 and inhibitor of FBPase-1, its reduction inhibits glycolysis and stimulates gluconeogenesis. Therefore, in response to glucagon, hepatic glucose production increases, helping the liver counteract the drop in blood glucose levels.[28][29]
Note: like adrenaline, glucagon also promotes gluconeogenesis by increasing the availability of key substrates such as glycerol and amino acids.[30]
Insulin
Insulin has the opposite effect. Upon receptor binding, it activates phosphoprotein phosphatase 2A (PP2A), which dephosphorylates PFK-2/FBPase-2:
- this activates PFK-2
- and inhibits FBPase-2
Insulin also stimulates cAMP phosphodiesterase, which degrades cAMP into AMP, further reducing PKA activity. The result is an increase in F2,6BP levels, which inhibits gluconeogenesis and stimulates glycolysis.
Additionally:
- fructose 6-phosphate allosterically activates PFK-2 and inhibits FBPase-2;
- both PFK-2 and FBPase-2 are subject to product inhibition.
However, the main regulatory factors are the level of fructose 6-phosphate and the phosphorylation state of the bifunctional enzyme.[31]
Glucose 6-phosphatase
Unlike pyruvate carboxylase and fructose-1,6-bisphosphatase, the catalytic subunit of glucose 6-phosphatase is not regulated allosterically or through covalent modification. Instead, its activity is modulated at the transcriptional level.[32]
Conditions that promote glucose production, such as low blood glucose, glucagon, and glucocorticoids, stimulate the expression of the enzyme. In contrast, insulin acts as a negative regulator, inhibiting its synthesis.[33]
Additionally, the Km of glucose 6-phosphatase for glucose 6-phosphate is significantly higher than the physiological concentration of the substrate. This means the enzyme’s activity is almost linearly dependent on the substrate concentration, making it effectively regulated by substrate availability rather than by allosteric effectors.[34]
PEP carboxykinase
PEP carboxykinase is expressed in both the cytosol and mitochondria of hepatocytes, encoded by separate nuclear genes. Its expression is tightly regulated, primarily through changes in its synthesis and degradation rates. For example, enzyme levels are low before birth but rise markedly within hours after delivery, coinciding with the activation of gluconeogenesis in the newborn.[35][36]
High levels of glucagon or fasting promote enzyme production by stabilizing its mRNA and enhancing gene transcription. Conversely, elevated blood glucose or insulin suppress transcription, thereby reducing enzyme levels.[34]
Xylulose 5-phosphate
Xylulose 5-phosphate, a product of the pentose phosphate pathway, is a relatively recently identified regulatory molecule. It stimulates glycolysis and inhibits gluconeogenesis by modulating the concentration of fructose 2,6-bisphosphate in the liver.[37]
When blood glucose levels rise, such as after a carbohydrate-rich meal, both glycolysis and the pentose phosphate pathway are activated in hepatocytes. The resulting production of xylulose 5-phosphate activates protein phosphatase 2A. As previously described, PP2A dephosphorylates PFK-2/FBPase-2, thereby inhibiting FBPase-2 and activating PFK-2. This leads to a rise in fructose 2,6-bisphosphate levels, which in turn inhibits gluconeogenesis and stimulates glycolysis. The increased glycolytic flux results in the production of acetyl-CoA, a key precursor for lipid synthesis.[38]
Simultaneously, the enhanced activity of the pentose phosphate pathway produces NADPH, which provides reducing power for fatty acid biosynthesis. Additionally, PP2A dephosphorylates carbohydrate-responsive element-binding protein (ChREBP), a transcription factor that upregulates the expression of hepatic genes involved in lipid synthesis.[39]
Thus, in response to elevated blood glucose levels, xylulose 5-phosphate acts as a crucial regulator, promoting lipid synthesis and coordinating carbohydrate and fat metabolism.[38]
Precursors
Besides the aforementioned pyruvate, the major gluconeogenic precursors are lactate, glycerol, most amino acids, and, more generally, any compound that can be converted into pyruvate or oxaloacetate.[40]
Glycerol
Glycerol is released by lipolysis in adipose tissue. With the exception of propionyl-CoA, it is the only part of the lipid molecule that can be used for glucose synthesis in animals.[5]
Glycerol enters gluconeogenesis, or glycolysis, depending on the cellular energy charge, as dihydroxyacetone phosphate (DHAP). Its synthesis occurs in two steps.
In the first step, glycerol is phosphorylated to glycerol 3-phosphate by the enzyme glycerol kinase (EC 2.7.1.30), with the consumption of one ATP.
Glycerol + ATP → Glycerol 3-phosphate + ADP
This enzyme is absent in adipocytes, but present in the liver. This means that glycerol must reach the liver to be further metabolized.
Glycerol 3-phosphate is then oxidized to DHAP in a reaction catalyzed by glycerol 3-phosphate dehydrogenase (EC 1.1.1.8), during which NAD+ is reduced to NADH.[9]
Glycerol 3-phosphate + NAD+ ⇄ DHAP + NADH + H+
During prolonged fasting, glycerol becomes the major gluconeogenic precursor, accounting for approximately 20% of total glucose production.[41]
Glucogenic amino acids
Pyruvate and oxaloacetate are the entry points for the glucogenic amino acids, that is, those whose carbon skeleton, or part of it, can be used for de novo glucose synthesis.
Amino acids are derived from the catabolism of proteins, whether dietary or endogenous, such as skeletal muscle proteins during fasting or intense, prolonged exercise.
The catabolic pathways of the twenty amino acids that make up proteins converge to form seven major metabolic products:
- acetyl-CoA
- acetoacetyl-CoA
- α-ketoglutarate
- succinyl-CoA
- fumarate
- oxaloacetate
- pyruvate.[21]
Among these, only acetyl-CoA and acetoacetyl-CoA cannot be used for gluconeogenesis. Therefore, glucogenic amino acids can be defined as those whose carbon skeleton (or a part of it) can be converted into pyruvate, oxaloacetate, α-ketoglutarate, succinyl-CoA, or fumarate.
Note: only leucine and lysine are exclusively ketogenic, as their carbon skeletons are broken down into acetyl-CoA and/or acetoacetyl-CoA, which cannot serve as gluconeogenic substrates.
Below are the entry points of the gluconeogenic amino acids.
- Pyruvate: alanine, cysteine, glycine, serine, threonine, tryptophan.
- Oxaloacetate: aspartate, asparagine.
- α-Ketoglutarate: glutamate, arginine, glutamine, histidine, proline.
- Succinyl-CoA: isoleucine, methionine, threonine, valine.
- Fumarate: phenylalanine, tyrosine.
The intermediates α-ketoglutarate, succinyl-CoA, and fumarate, all part of the citric acid cycle, enter the gluconeogenic pathway after conversion to oxaloacetate.[42]
Propionate
Propionate, a short-chain fatty acid, is a gluconeogenic precursor because its active form, propionyl-CoA, can be converted into succinyl-CoA.
It originates from several sources.
- β-Oxidation of odd-chain fatty acids, such as margaric acid (C17:0). In the final β-oxidation step, a five-carbon fatty acid is split into acetyl-CoA and propionyl-CoA. These fatty acids, though less common, are found in lipids of marine organisms, ruminants, and some plants.[43]
- Oxidation of branched-chain fatty acids, like phytanic acid, derived from phytol, a chlorophyll breakdown product.[44]
- In ruminants, microbial fermentation of cellulose-derived glucose in the rumen produces propionate. Once absorbed, it can be used for gluconeogenesis, fatty acid synthesis, or energy production. In these animals, where gluconeogenesis is nearly constant, propionate is the major gluconeogenic precursor.[45]
- It also derives from the catabolism of the amino acids valine, isoleucine, methionine, and threonine.[46]
- In the colon, fermentation of non-digestible carbohydrates (e.g., resistant starch and fibers) by the gut microbiota produces propionate. After absorption by enterocytes, the portion not metabolized locally reaches the liver via the portal vein.[47]
Conversion of propionyl-CoA to succinyl-CoA involves three reactions:
- Propionyl-CoA carboxylase (EC 6.4.1.3, biotin-dependent) catalyzes:
Propionyl-CoA + HCO3− + ATP → D-methylmalonyl-CoA + ADP + Pi
Methylmalonyl-CoA epimerase (EC 5.1.99.1) converts D- to L-methylmalonyl-CoA.
D-Methylmalonyl-CoA ⇄ L-Methylmalonyl-CoA
Methylmalonyl-CoA mutase (EC 5.4.99.2), requiring coenzyme B12, rearranges the molecule to succinyl-CoA.[5]
L-Methylmalonyl-CoA ⇄ Succinyl-CoA
References
- ^ a b c d e f g Garrett R.H., Grisham C.M. Biochemistry. 7th Edition. Cengage Learning, 2023.
- ^ a b c d e f g Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
- ^ a b c d e Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
- ^ a b c d e f Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 4th Edition. Elsevier health sciences, 2018.
- ^ a b c d e Chourpiliadis C., Mohiuddin S.S. Biochemistry, Gluconeogenesis. [Updated 2023 Jun 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK544346/
- ^ a b Cahill G.F. Jr. Fuel metabolism in starvation. Annu Rev Nutr 2006;26:1-22. doi:10.1146/annurev.nutr.26.061505.111258
- ^ Cryer P.E. Hypoglycemia, functional brain failure, and brain death. J Clin Invest 2007;117(4):868-70. doi:10.1172/JCI31669
- ^ a b Gerich J.E. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med 2010;27(2):136-42. doi:10.1111/j.1464-5491.2009.02894.x
- ^ a b c Melkonian E.A., Asuka E., Schury M.P. Physiology, Gluconeogenesis. [Updated 2023 Nov 13]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK541119/
- ^ Adina-Zada A., Zeczycki T.N., Attwood P.V. Regulation of the structure and activity of pyruvate carboxylase by acetyl CoA. Arch Biochem Biophys 2012;519(2):118-30. doi:10.1016/j.abb.2011.11.015
- ^ Utter M.F., Keech D.B. Formation of oxaloacetate from pyruvate and carbon dioxide. J Biol Chem 1960;235:PC17-8. doi:10.1016/S0021-9258(18)69442-6
- ^ Case C.L., Mukhopadhyay B. Kinetic characterization of recombinant human cytosolic phosphoenolpyruvate carboxykinase with and without a His10-tag. Biochim Biophys Acta 2007;1770(11):1576-84. doi:10.1016/j.bbagen.2007.07.012
- ^ McCommis K.S. and Finck B.N. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J 2015;466(3):443-454. doi:10.1042/BJ20141171
- ^ Wu G. Amino acids: biochemistry and nutrition. CRC Press, 2010.
- ^ Holeček M. Roles of malate and aspartate in gluconeogenesis in various physiological and pathological states. Metabolism 2023;145:155614. doi:10.1016/j.metabol.2023.155614
- ^ American Chemical Society National Historic Chemical Landmarks. Carl and Gerty Cori and Carbohydrate Metabolism. https://www.acs.org/education/whatischemistry/landmarks/carbohydratemetabolism.html. Accessed July 6, 2025.
- ^ a b c van Schaftingen E., Gerin I. The glucose-6-phosphatase system. Biochem J 2002;362(Pt 3):513-32. doi:10.1042/0264-6021:3620513
- ^ Newsholme E.A. Substrate cycles: their metabolic, energetic and thermic consequences in man. Biochem Soc Symp 1978;43:183-205.
- ^ Patel M.S., Korotchkina L.G. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans 2006;34(2):217-22. doi:10.1042/bst0340217
- ^ Jitrapakdee S., St Maurice M., Rayment I., Cleland W.W., Wallace J.C., Attwood P.V. Structure, mechanism and regulation of pyruvate carboxylase. Biochem J 2008;413(3):369-87. doi:10.1042/BJ20080709
- ^ a b c d Rosenthal M.D., Glew R.H. Medical biochemistry – Human metabolism in health and disease. John Wiley & Sons, Inc., Publication, 2009.
- ^ Villeret V., Huang S., Zhang Y., Lipscomb W.N. Structural aspects of the allosteric inhibition of fructose-1,6-bisphosphatase by AMP: the binding of both the substrate analogue 2,5-anhydro-D-glucitol 1,6-bisphosphate and catalytic metal ions monitored by X-ray crystallography. Biochemistry 1995;34(13):4307-15. doi:10.1021/bi00013a020
- ^ Lynch E.M., Hansen H., Salay L., Cooper M., Timr S., Kollman J.M., Webb B.A. Structural basis for allosteric regulation of human phosphofructokinase-1. Nat Commun 2024;15(1):7323. doi:10.1038/s41467-024-51808-6
- ^ Rui L. Energy metabolism in the liver. Compr Physiol 2014;4(1):177-97. doi:10.1002/cphy.c130024
- ^ Van Schaftingen E., and Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:https://doi.org/10.1073/pnas.78.5.2861
- ^ Van Schaftingen E., Jett M-F., Hue L., and Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:https://doi.org/10.1073/pnas.78.6.3483
- ^ Okar DA., Lange A.J. Fructose-2,6-bisphosphate and control of carbohydrate metabolism in eukaryotes. Biofactors 1999;10(1):1-14. doi:https://doi.org/10.1002/biof.5520100101
- ^ Agius L. Hormonal and metabolite regulation of hepatic glucokinase. Annu Rev Nutr 2016;36:389-415. doi:https://doi.org/10.1146/annurev-nutr-071715-051145
- ^ Payne V.A., Arden C., Wu C., Lange A.J., Agius L. Dual role of phosphofructokinase-2/fructose bisphosphatase-2 in regulating the compartmentation and expression of glucokinase in hepatocytes. Diabetes 2005;54(7):1949-57. doi:https://doi.org/10.2337/diabetes.54.7.1949
- ^ Winther-Sørensen M., Galsgaard K.D., Santos A., et al. Glucagon acutely regulates hepatic amino acid catabolism and the effect may be disturbed by steatosis. Mol Metab 2020;42:101080. doi:https://doi.org/10.1016/j.molmet.2020.101080
- ^ Hatting M., Tavares C.D.J., Sharabi K., Rines A.K., Puigserver P. Insulin regulation of gluconeogenesis. Ann N Y Acad Sci 2018;1411(1):21-35. doi:https://doi.org/10.1111/nyas.13435
- ^ Soty M., Chilloux J., Delalande F., Zitoun C., Bertile F., Mithieux G., and Gautier-Stein A. Post-Translational regulation of the glucose-6-phosphatase complex by cyclic adenosine monophosphate is a crucial determinant of endogenous glucose production and is controlled by the glucose-6-phosphate transporter. J Proteome Res 2016;15(4):1342-1349. doi:https://doi.org/10.1021/acs.jproteome.6b00110
- ^ Schmoll D., Walker K.S., Alessi D.R., Grempler R., Burchell A., Guo S., Walther R., Unterman T.G. Regulation of glucose-6-phosphatase gene expression by protein kinase Balpha and the forkhead transcription factor FKHR. Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity. J Biol Chem 2000;275(46):36324-33. doi:https://doi.org/10.1074/jbc.M003616200
- ^ a b Rodwell V.W., Bender D.A., Botham K.M., Kennelly P.J., Weil P.A. Harper’s Illustrated Biochemistry. 31st Edition. McGraw-Hill, 2018.
- ^ Hanson R.W., Reshef L. Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev Biochem 1997;66:581-611. doi:https://doi.org/10.1146/annurev-biochem.66.1.581
- ^ Yabaluri N., Bashyam M.D. Hormonal regulation of gluconeogenic gene transcription in the liver. J Biosci 2010;35(3):473-84. doi:https://doi.org/10.1007/s12038-010-0052-0
- ^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:https://doi.org/10.1073/pnas.0730817100
- ^ a b Uyeda K. Short- and long-term adaptation to altered levels of glucose: fifty years of scientific adventure. Annu Rev Biochem 2021;90:31-55. doi:https://doi.org/10.1146/annurev-biochem-070820-125228
- ^ Iizuka K., Takao K., Yabe D. ChREBP-mediated regulation of lipid metabolism: involvement of the gut microbiota, liver, and adipose tissue. Front Endocrinol (Lausanne) 2020;11:587189. doi:https://doi.org/10.3389/fendo.2020.587189
- ^ Shah A.M., Wondisford F.E. Tracking the carbons supplying gluconeogenesis. J Biol Chem 2020;295(42):14419-14429. doi:https://doi.org/10.1074/jbc.rev120.012758
- ^ Baba H., Zhang X.J., Wolfe R.R. Glycerol gluconeogenesis in fasting humans. Nutrition 1995;11(2):149-53.
- ^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
- ^ Ferrannini E., Barrett E.J., Bevilacqua S., DeFronzo R.A. Effect of fatty acids on glucose production and utilization in man. J Clin Invest 1983;72(5):1737-47. doi:https://doi.org/10.1172/JCI111133
- ^ Verhoeven N.M., Wanders R.J., Poll-The B.T., Saudubray J.M., Jakobs C. The metabolism of phytanic acid and pristanic acid in man: a review. J Inherit Metab Dis 1998;21(7):697-728. doi:https://doi.org/10.1023/a:1005476631419
- ^ Short-chain fatty acids regulate intestinal gluconeogenesis. Nat Rev Endocrinol 2014;10:190 (2014). doi:https://doi.org/10.1038/nrendo.2014.9
- ^ Mann G., Mora S., Madu G., Adegoke O.A.J. Branched-chain amino acids: catabolism in skeletal muscle and implications for muscle and whole-body metabolism. Front Physiol 2021;12:702826. doi:https://doi.org/10.3389/fphys.2021.702826
- ^ Portincasa P., Bonfrate L., Vacca M., De Angelis M., Farella I., Lanza E., Khalil M., Wang D.Q.-H., Sperandio M., Di Ciaula A. Gut microbiota and short chain fatty acids: implications in glucose homeostasis. Int J Mol Sci 2022;23:1105. doi:https://doi.org/10.3390/ijms23031105