Flavonoid biosynthesis, probably the best characterized pathway of plant secondary metabolism, is part of the phenylpropanoid pathway that, in addition to flavonoids, leads to the formation of a wide range of phenolic compounds, such as hydroxycinnamic acids, stilbenes, lignans and lignins.
Flavonoid biosynthesis is linked to primary metabolism through both mitochondria- and plastid-derived molecules. Since it seems that most of the involved enzymes characterized to date operate in protein complexes located in the cell cytosol, these molecules must be exported to the cytoplasm to be used.
The end products are transported to different intracellular or extracellular locations, with flavonoids involved in pigmentation usually transported into the vacuoles.
The biosynthesis of this group of polyphenols requires one p-coumaroyl-CoA and three malonyl-CoA molecules as initial substrates.
Contents
- Biosynthesis of p-coumaroyl-CoA
- Biosynthesis of malonyl-CoA
- First steps in flavonoid biosynthesis
- References
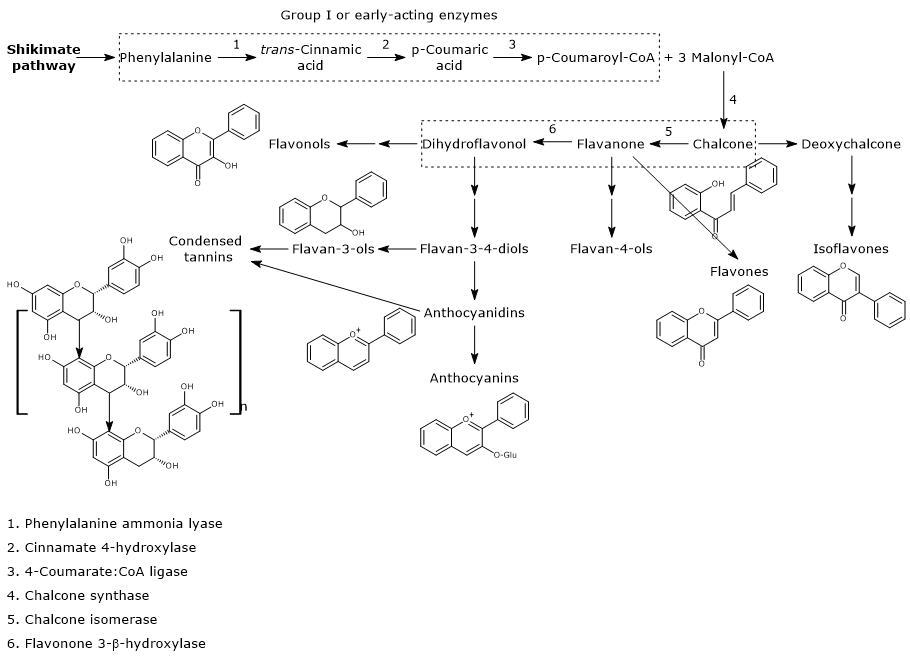
Biosynthesis of p-coumaroyl-CoA
p-Coumaroyl-CoA is the pivotal branch-point metabolite in the phenylpropanoid pathway, being the precursor of a wide variety of phenolic compounds, both flavonoid and non-flavonoid polyphenols.
It is produced from phenylalanine via three reactions catalyzed by cytosolic enzymes collectively called group I or early-acting enzymes, in order of action:
- phenylalanine ammonia lyase (EC 4.3.1.24);
- trans-cinnamate 4-monooxygenase (EC:1.14.14.91);
- 4-coumarate-CoA ligase (EC 6.2.1.12).
They seems to be associated in a multienzyme complex anchored to the endoplasmic reticulum membrane. The anchoring is probably ensured by cinnamate 4-hydroxylase that inserts its N-terminal domain into the membrane of the endoplasmic reticulum itself. These complexes, referred to as “metabolons”, allow the product of a reaction to be channeled directly as substrate to the active site of the enzyme that catalyzes the consecutive reaction in the metabolic pathway.
With the exception of cinnamate 4-hydroxylase, the enzymes which act downstream of phenylalanine ammonia lyase are encoded by small gene families in all species analyzed so far.
The different isoenzymes show distinct temporal, tissue, and elicitor-induced patterns of expression. It seems, in fact, that each member of each family can be used mainly for the synthesis of a specific compound, thus acting as a control point for carbon flux among the metabolic pathways leading to lignan, lignin, and flavonoid biosynthesis.
Note: Phenylalanine is a product of the shikimic acid pathway, which converts simple precursors derived from the metabolism of carbohydrates, phosphoenolpyruvate and erythrose-4-phosphate, intermediates of glycolysis and the pentose phosphate pathway, respectively, into the aromatic amino acids phenylalanine, tyrosine and tryptophan. Unlike plants and microorganisms, animals do not possess the shikimic acid pathway, and are not able to synthesize the three above-mentioned amino acids, which are therefore essential nutrients.
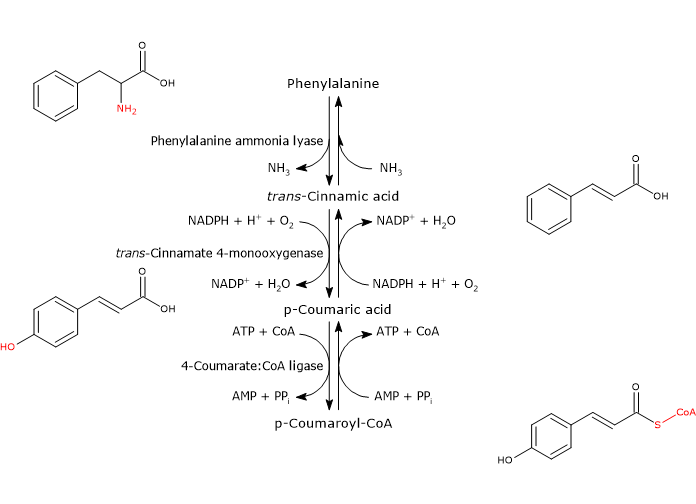
Phenylalanine ammonia lyase (PAL)
It is one of the most studied and best characterized enzymes of plant secondary metabolism. It requires no cofactors and catalyzes the reaction that links primary and secondary metabolism: the reversible deamination of phenylalanine to trans-cinnamic acid, with the release of nitrogen as ammonia and introduction of a trans double bond between carbon atoms 7 and 8 of the side chain.
Phenylalanine ⇄ trans-Cinnamic Acid + NH3
Therefore, it directs the flow of carbon from the shikimic acid pathway to the different branches of the phenylpropanoid pathway. The released ammonia is probably fixed in the reaction catalyzed by glutamine synthetase.
The enzyme from monocots is also able to act as tyrosine ammonia lyase (EC 4.3.1.25), converting tyrosine to p-coumaric acid directly, (therefore without the 4-hydroxylation step), but with a lower efficiency.
In all plant species investigated, several copies of phenylalanine ammonia lyase gene are found, copies that probably respond differentially to internal and external stimuli. Indeed, gene transcription, and then enzyme activity, are under the control of both internal developmental and external environmental stimuli.
Here are some examples that require increased enzyme activity.
- The flowering.
- The production of lignin to strengthen the secondary wall of xylem cells.
- The production of flower pigments that attract pollinators.
- Pathogen infections, that require the production of phenylpropanoid phytoalexins, or exposure to UV rays.
trans-Cinnamate 4-monooxygenase
It belongs to the cytochrome P450 superfamily (EC 1.14.-.-), is a microsomal monooxygenase containing a heme cofactor, and dependent on both O2 and NADPH.
It catalyzes the formation of p-coumaric acid through the introduction of a hydroxyl group in 4-position of trans-cinnamic acid (this hydroxyl group is present in most flavonoids).
trans-Cinnamic Acid + NADPH + H+ + O2 ⇄ p-Coumaric Acid + NADP+ + H2O
This reaction is also part of the biosynthesis of hydroxycinnamic acids.
Increases in transcription rates and enzyme activity are observed in correlation with the synthesis of phytoalexins (in response to fungal infections), lignification as well as wounding.
4-Coumarate:CoA ligase (4CL)
With Mg2+ as a cofactor, it catalyzes the ATP-dependent activation of the carboxyl group of p-coumaric acid and other hydroxycinnamic acids, metabolically rather inert molecules, through the formation of the corresponding CoA-thioester.
p-Coumaric Acid + ATP + CoA ⇄ p-Coumaroyl-CoA + AMP + PPi
Generally, p-coumaric acid and caffeic acid are the preferred substrates, followed by ferulic acid and 5-hydroxyferulic acid, low activity against trans-cinnamic acid and none against sinapic acid. These CoA-thioesters are able to enter various reactions such as:
- reduction to alcohol (monolignols) or aldehydes;
- stilbene and flavonoid biosynthesis;
- transfer to acceptor molecules.
It should finally be pointed out that the activation of the carboxyl group can also be obtained through an UDP-glucose-dependent transfer to glucose.
Biosynthesis of malonyl-CoA
Malonyl-CoA does not derived from the phenylpropanoid pathway, but from the reaction catalyzed by acetyl-CoA carboxylase (EC 6.4.1.2, the cytosolic form, see below).
The enzyme, with biotin and Mg2+ as cofactors, catalyzes the ATP-dependent carboxylation of acetyl-CoA, using bicarbonate as a source of carbon dioxide (CO2).
Acetyl-CoA + HCO3– + ATP → Malonyl-CoA + ADP + Pi
It is found both in the plastids, where it participates in the synthesis of fatty acids, and the cytoplasm, and is the latter that catalyzes the formation of malonyl-CoA that is used in the biosynthesis of flavonoids and other compounds.
Increases in the transcription rate of the gene and enzyme activity are induced in response to stimuli that increase the biosynthesis of these polyphenols, such as exposure to pathogenic fungi or UV-rays.
In turn, acetyl-CoA is produced in plastids, mitochondria, peroxisomes and cytosol through different metabolic pathways. The molecules used in the biosynthesis of malonyl-CoA, and therefore of the flavonoids, are the cytosolic ones, produced in the reaction catalyzed by ATP-citrate lyase (EC 2.3.3.8) that cleaves citrate, in the presence of CoA and ATP, to form oxaloacetate and acetyl-CoA, plus ADP and inorganic phosphate.
First steps in flavonoid biosynthesis
The first step in flavonoid biosynthesis is catalyzed by chalcone synthase (EC 2.3.1.74), an enzyme anchored to the endoplasmic reticulum and with no known cofactors.
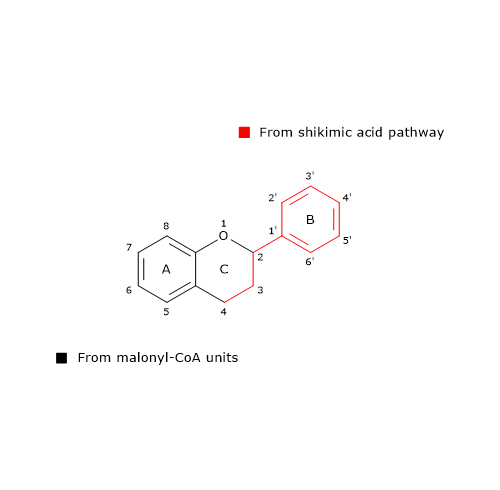
From one p-coumaroyl-CoA and three malonyl-CoA, it catalyzes sequential condensation and decarboxylation reactions in the course of which a polyketide intermediate is formed. The polyketide undergoes cyclizations and aromatizations leading to the formation of the A ring. The product of the reactions is naringenin chalcone (2′,4,4′,6′-tetrahydroxychalcone), a 6′-hydroxychalcone and the first flavonoid to be synthesized by plants.
p-Coumaroyl-CoA + 3 Malonyl-CoA → Naringenin Chalcone + 4 CoA + 3 CO2
The reaction, cytosolic, is irreversible due to the release of three CO2 and 4 CoA.
B ring and the three-carbon bridge of the molecule originate from p-coumaroyl-CoA (and therefore from phenylalanine), the A ring from the three malonyl-CoA units.
Also 6’-deoxychalcone can be produced; its synthesis is thought to involve an additional reduction step catalyzed by polyketide reductase (EC. 1.1.1.-).
Chalcone synthase from some plant species, such as barley (Hordeum vulgare), accepts as substrates also caffeoil-CoA, feruloil-CoA and cinnamoyl-CoA. It is the most abundant enzyme of the phenylpropanoid pathway, probably because it has a low catalytic activity, and, in fact, is considered to be the rate-limiting enzyme in flavonoid biosynthesis.
As for phenylalanine ammonia lyase, chalcone synthase gene expression is under the control of both internal and external factors. In some plants, one or two isoenzymes are found, while in others up to 9.
Chalcone synthase belongs to polyketide synthase group, present in bacteria, fungi and plants. These enzymes are able to catalyze the production of polyketide chains through sequential condensations of acetate units provided by malonyl-CoA units. They also includes stilbene synthase (EC 2.3.1.146), which catalyzes the formation of resveratrol, a non flavonoid polyphenol compound that has attracted much interest because of its potential health benefits.
Generally, chalcones do not accumulate in plants because naringenin chalcone is converted to (2S)-naringenin, a flavanone, in the reaction catalyzed by chalcone isomerase (EC 5.5.1.6).
The enzyme, the first of the flavonoid biosynthesis to be discovered, catalyzes a stereospecific isomerization and closes the C ring. Two types of chalcone isomerases are known, called type I and II. Type I enzymes use only 6′-hydroxychalcone substrates, like naringenin chalcone, while type II, prevalent in legumes, use both 6′-hydroxy- and 6′-deoxychalcone substrates.
It should be noted that with 6′-hydroxychalcones, isomerization can also occur nonenzymically to form a racemic mixture, both in vitro and in vivo, enough to allow a moderate synthesis of anthocyanins. On the contrary, under physiological conditions 6′-deoxychalcones are stable, and so the activity of type II chalcone isomerases is required to form flavanones.
The enzyme increases the rate of the reaction of 107 fold compared to the spontaneous reaction, but with a higher kinetics for the 6′-hydroxychalcones than 6′-deoxychalcones. Finally, it produces (2S)-flavanones, which are the biosynthetically required enantiomers.
As other enzymes in flavonoid biosynthesis, also chalcone isomerase gene expression is subject to strict control. And, as phenylalanine ammonia lyase and chalcone synthase, it is induced by elicitors.
In the reaction catalysed by flavanone-3β-hydroxylase (EC 1.14.11.9), (2S)-flavanones undergo a stereospecific isomerization that converts them into the respective (2R,3R)-dihydroflavonols. In particular, naringenin is converted into dihydrokaempferol.
The enzyme is a cytosolic non-heme-dependent dioxygenase, dependent on Fe2+ and 2-oxoglutarate, and therefore belonging to the family of 2-oxoglutarate-dependent dioxygenase (which distinguishes them from the other hydroxylases of the flavonoid biosynthetic pathway which are cytochrome P450 enzymes).
Naringenin chalcone, (2S)-naringenin, and dihydrokaempferol are central intermediates in flavonoid biosynthesis, since they act as branch-point compounds from which the synthesis of distinct flavonoid subclasses can occur. For example, directly or indirectly:
- isoflavones derive from chalcones;
- flavones and flavan-4-ols from flavanones;
- flavan-3,4-diols, anthocyanidins, anthocyanins, catechins, flavonols, and proanthocyanidins or condensed tannins from dihydroflavonols.
Not all of these side metabolic pathways are present in every plant species, or are active within each tissue type of a given plant.
Like enzymes previously seen, the activity of those involved in these “side-routes” is subject to strict control, resulting in a tissue-specific profile of flavonoid compounds. For example, grape seeds, flesh and skin have distinct anthocyanin, catechin, flavonol and condensed tannin profiles, whose synthesis and accumulation are strictly and temporally coordinated during the ripening process.
References
- Andersen Ø.M., Markham K.R. Flavonoids: chemistry, biochemistry, and applications. CRC Press Taylor & Francis Group, 2006
- de la Rosa L.A., Alvarez-Parrilla E., Gonzàlez-Aguilar G.A. Fruit and vegetable phytochemicals: chemistry, nutritional value, and stability. 1th Edition. Wiley J. & Sons, Inc., Publication, 2010
- Heldt H-W. Plant biochemistry – 3th Edition. Elsevier Academic Press, 2005
- Vogt T. Phenylpropanoid biosynthesis. Mol Plant 2010;3(1):2-20. doi:10.1093/mp/ssp106
- Wink M. Biochemistry of plant secondary metabolism – 2nd Edition. Annual plant reviews (v. 40), Wiley J. & Sons, Inc., Publication, 2010