Glycogen is a branched polymer composed of D-glucose units and serves as a storage form of the monosaccharide, and therefore of energy.[1]
As it is synthesized without a template, unlike proteins and nucleic acids, it exists as a population of molecules with different structures and sizes.[2][3]
Due to its branched structure, glycogen is a compact and soluble macromolecule that exerts low osmotic pressure and allows rapid release of stored glucose when needed.[4]
The glycogen molecule forms assemblages with proteins essential for its metabolism, such as glycogenin (EC 2.4.1.186), glycogen synthase (EC 2.4.1.11), glycogen phosphorylase (EC 2.4.1.1), and the debranching enzyme (EC 2.4.1.25 and EC 3.2.1.33), as well as others that regulate its anchoring to the cytoskeleton and membranes. Such polysaccharide–protein aggregates are called β-granules and are found in the cytosol of bacteria, Archaea, fungi, and animal cells. Furthermore, phosphate groups are covalently linked to the polysaccharide.[5]
Proteins and phosphate groups are involved in the regulation of its metabolism, which, as with metabolism in general, consists of two phases: catabolism and anabolism. During the anabolic phase, reactions catalyzed by specific enzymes lead to glycogen synthesis, whereas during the catabolic phase, reactions catalyzed by specific enzymes lead to glycogen breakdown, or glycogenolysis.[6]
In animals, glycogen is found in practically all cells and, in mammals, it is most abundant in the liver and skeletal muscle. Within the liver, several β-granules assemble to form the so-called α-granules.[3] Glycogen is also found in lysosomes.[7]
In humans, it represents less than 1% of the body’s energy stores and is essential for maintaining blood glucose homeostasis.[8]
It is absent in plants, where starch serves as the storage form of glucose. Therefore, the polymerization of glucose represents a universal mechanism for energy storage.[9]
Glycogen has little nutritional significance for humans.[10]
Contents
- Historical background
- Chemical and molecular structure of glycogen
- β-Granules
- α-Granules
- Glycogen metabolism: the synthetic phase
- Glycogen metabolism: the degradative phase
- Coordinated regulation of glycogen metabolism
- Localization of glycogen in humans
- Why it is important to humans
- Glycogen and muscle work
- Energy yield under anaerobic conditions
- Energy yield under aerobic conditions
- References
Historical background
Glycogen was discovered in 1857 by the French physiologist Claude Bernard, who is considered the founder of experimental medicine.[11] In the second half of the 20th century, studies on glycogen metabolism led to several significant discoveries such as:
- the reversible phosphorylation of proteins;
- protein kinases and protein phosphatases;
- the effect of insulin on the activity of intracellular enzymes.[9]
In turn, this led to the award of four Nobel Prizes, three for Physiology or Medicine, to Carl Ferdinand Cori and Gerty Theresa Cori (née Radnitz) in 1947, Earl Sutherland Jr. in 1971, and Edwin Krebs and Edmond Fischer in 1992, and one for Chemistry, to Louis Leloir in 1970.[12]
Chemical and molecular structure of glycogen
An individual glycogen molecule is a branched polymer of D-glucose in the pyranose form, that is, a rigid six-membered heterocyclic ring composed of five carbon atoms and one oxygen atom, with a chair conformation.
The central priming protein glycogenin and phosphate groups are covalently bound to the polysaccharide chain.[1]
Most of the glucose units are linked by α-1,4-glycosidic bonds, where each unit is connected to the next through a bond between the C-1 atom of one unit and the hydroxyl group on the C-4 atom of the next, with an oxygen atom acting as a bridge between the two carbons.
Branch points are introduced by α-1,6-glycosidic bonds, which occur approximately every 8–12 residues, again with an oxygen atom bridging the two carbons (C-1 and C-6), and with an average chain length of about 13 residues in mammals. Because each branch terminates in a non-reducing residue, there are n + 1 non-reducing ends in the molecule, where n is the number of chains, but only one reducing end, to which glycogenin is linked.[13]
Note: in disaccharides, oligosaccharides, and polysaccharides, the non-reducing end is the end that lacks a free anomeric carbon atom.
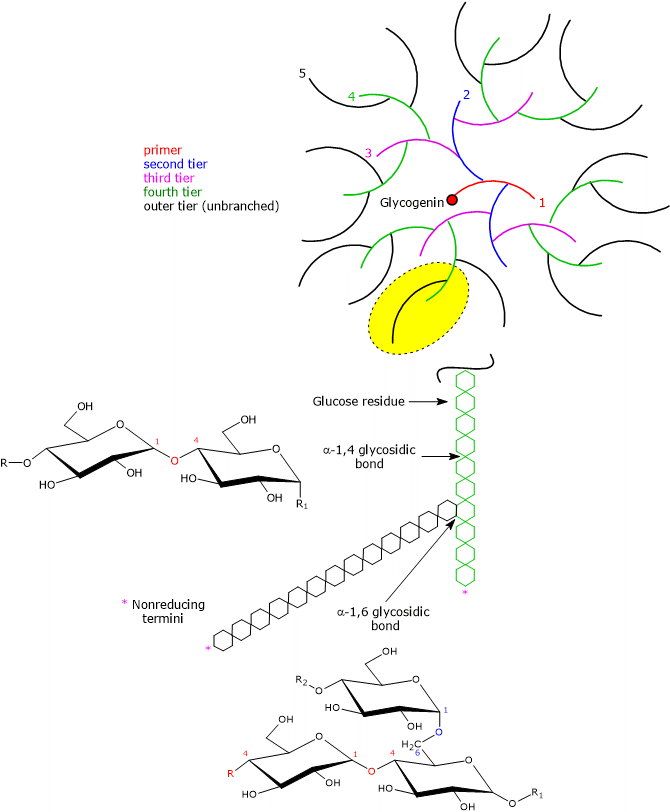
Having the same types of bonds, the primary structure of glycogen resembles that of amylopectin, which, together with amylose, is one of the two polymers of D-glucose units composing starch. However, compared with amylopectin, where branches occur every 25–30 glucose units, glycogen is more highly branched, and the branches are shorter.[14]
Heterogeneity of glycogen structure and particle size
Unlike proteins and nucleic acids, polysaccharides are synthesized without a template, through the addition of monosaccharides or oligosaccharides to the growing chain. Furthermore, because branching occurs without precise localization, molecules with the same mass do not necessarily share the same structure. Hence, for each type of molecule, there are multiple chemical structures.[3]
Moreover, glycogen isolated from different biological sources exists as a population of molecules with different sizes. Therefore, the best way to describe its chemistry is to define the distribution of molecular masses, as well as the average frequency and length of branches.[2]
Finally, it should be emphasized that glycogen is not a static entity but constantly varies over the course of its existence.[5]
As glucose has chiral centers, it exists as a pair of enantiomers, designated according to the Fischer-Rosanoff convention as D-glucose, the most widespread in nature and the monomeric unit of glycogen and starch, and L-glucose.[15]
Factors stabilizing the 3D structure
The folding of macromolecules such as proteins, nucleic acids, and polysaccharides into three-dimensional structures is governed by the same principles. The monomeric units, namely amino acids, nucleotides, and monosaccharides, with their more or less rigid structures, are joined by covalent bonds to form one-dimensional polymers that spontaneously fold into three-dimensional conformations stabilized by noncovalent interactions such as:
- hydrogen bonds;
- van der Waals interactions;
- hydrophobic interactions;
- ionic interactions, when charged subunits are present.[13]
These interactions can occur within macromolecules or between macromolecules, as in supramolecular complexes such as cellulose or multienzyme complexes.[16]
Because the pyranose ring of glucose is a rigid structure, the three-dimensional conformation of oligosaccharides and polysaccharides results from rotation about both C–O bonds of the glycosidic linkage, with the bond angles denoted as Φ (phi) and Ψ (psi). However, there is no free rotation about each C–O bond due to steric interference from substituents. Hence, some conformations are more stable than others. For amylose and glycogen, the most stable 3D structure is a tightly coiled helix stabilized by interchain hydrogen bonds.[9][17]
Advantages of the branched structure
The highly branched structure of glycogen offers several advantages.
- The non-reducing ends present on the outermost tier can act as substrates for glycogen phosphorylase. Therefore, many glycogen phosphorylase molecules can work simultaneously, allowing rapid mobilization of stored glucose as glucose 1-phosphate (G1P).[14]
- The highly branched structure also allows stored glucose to exert a much lower osmotic pressure than it would if present in its monomeric form. For example, hepatocytes store an amount of glucose that, if free, would correspond to a concentration of about 0.4 M, compared with a glycogen concentration of about 0.01 mM. Therefore, if glucose were in free form, the resulting osmolarity would be so high that it would cause an osmotic influx of water leading to cell lysis. Moreover, since the extracellular concentration of glucose is about 5 mM, glucose uptake into a cell with an internal glucose concentration of 0.4 M would be particularly energy-expensive.[9]
- Branches also allow the formation of compact granules.
- If branches were absent or few, a very large number of long linear polymers would have to be present to provide a comparable number of non-reducing ends and to store a similar amount of glucose. This could cause cell damage. Evidence supporting this hypothesis comes from a rare genetic disorder, Andersen’s disease (also called amylopectinosis or glycogen storage disease type IV), caused by mutations in the gene encoding the branching enzyme. These mutations result in a deficiency of enzyme activity and in the accumulation, in various tissues, of abnormally branched glycogen resembling amylopectin.[2][6]
- Finally, branches allow glycogen to remain soluble, unlike starch.[5]
Whelan’s model
Due to the action of glycogenin, followed by glycogen synthase and the branching enzyme, also called glycosyl-(4,6)-transferase (EC 2.4.1.18), the glycogen molecule grows exponentially in concentric tiers around the glycogenin core. According to Whelan’s model of glycogen structure, two types of glucose chains can be identified:
- A-chains, which are unbranched and present only on the surface;
- B-chains, which are internal and contain, on average, two branching points.[18][19]
It has been calculated that the maximum size would be 12 tiers, with a diameter of about 42 nm, a total of approximately 55,000 glucose units, and a molecular mass of about 107 Da.[20][21]
Moreover, considering that each tier has a thickness of 3.8 nm, and assuming the glycogen molecule to be spherical, from the third to the twelfth tier:
- the diameter increases by a factor of 5.4;
- the volume, which grows according to the cube of the radius, increases 156-fold;
- the carbohydrate content increases 45.6 fold;
- the number of A-chains in each outermost tier increases exponentially and equals 2n − 1, where n corresponds to the number of the tier.[18]
| Level | Diameter (nm) | Chains/Level | Glucose/Level | Total Glucose |
|---|---|---|---|---|
| 1 | — | 1 | 13 | 13 |
| 2 | 3.8 | 3 | 26 | 39 |
| 3 | 7.8 | 7 | 52 | 91 |
| 4 | 11.6 | 15 | 104 | 195 |
| 5 | 15.4 | 31 | 208 | 403 |
| 6 | 19.2 | 63 | 416 | 819 |
| 7 | 23.0 | 127 | 832 | 1,651 |
| 8 | 26.8 | 255 | 1,664 | 3,315 |
| 9 | 30.6 | 511 | 3,328 | 6,643 |
| 10 | 34.4 | 1,023 | 6,656 | 13,299 |
| 11 | 38.2 | 2,047 | 13,312 | 26,611 |
| 12 | 42.0 | 4,095 | 26,624 | 53,235 |
In skeletal muscle, analysis of glycogen particle size by electron microscopy has shown the presence of only a few full-sized particles, with an average diameter of about 25 nm, corresponding to seven tiers.[22]
Feature of Whelan’s model
An important feature of Whelan’s model is that the outermost tier contains, in the form of A-chains, about 50% of all glucose residues. This does not mean that all of these molecules are accessible to glycogen phosphorylase, since the enzyme stops four residues away from the branch point. The intervention of the debranching enzyme, whose activity is slower than that of glycogen phosphorylase, removes the branch and allows glycogenolysis to proceed.[2][3]
Why the 13th tier is not possible
The 13th tier is considered impossible because of steric hindrance resulting from the high density of glucose units on the molecular surface. Such crowding would leave insufficient space for the interaction between the catalytic regions of enzymes involved in glycogen metabolism, including glycogen synthase, and the growing chains.[2][23]
Furthermore, mathematical analyses have suggested that the observed parameters, that is, branch length (about 13 residues in mammals), average branching frequency per tier (around 2), and the maximum number of tiers (12), are optimal for mobilizing the greatest possible number of glucose molecules in the shortest possible time.[24]
Glycogenin
The structure of the glycogen molecule includes the protein glycogenin, which is covalently bound to the polysaccharide chain. Glycogenin initiates glycogen synthesis by autoglycosylation, catalyzing the addition of 7–11 glucose units to a specific tyrosine residue. This primer chain then serves as a substrate for glycogen synthase. In addition, by binding to actin filaments, glycogenin anchors the oligosaccharide primer chain to the cytoskeleton.[14][25]
Phosphate groups
In addition to glycogenin, the glycogen molecule covalently binds phosphate groups.
For many years, these were considered contaminants, and their amounts were thought to be inversely correlated with the purity of the sample. Only in the early 1980s were they recognized as integral components of the polysaccharide, where they appear to be linked to C-2 and C-3 as monoesters, probably as a result of a side reaction during the activity of glycogen synthase.[26]
Many studies have suggested that their presence plays a role in regulating glycogen metabolism, similarly to what occurs in starch metabolism in plants. Evidence supporting this hypothesis includes the identification of laforin, a glycogen phosphatase, and the fact that mutations in its gene are a key factor in Lafora disease, a form of epilepsy characterized, among other things, by excessive phosphorylation of glycogen.[27]
But how do phosphate groups act? Several hypotheses have been proposed, and two are reported below.
- It has been suggested that phosphate groups, which are hydrophilic, could expose hydrophobic regions, thereby reducing glycogen solubility. Dephosphorylation by laforin, by maintaining polysaccharide solubility, would facilitate branch formation.[28]
- Another hypothesis suggests that the degree of phosphorylation may be related to the age of the molecule, acting as a kind of quality control. The increase in phosphorylation, which reduces the solubility of the polysaccharide, could serve as a metabolic marker that directs it toward lysosomal degradation, a process known as glycophagy, rather than toward glycogenolysis.[29]
β-Granules
Individual glycogen molecules are too small to be detected by light microscopy. Conversely, electron microscopy has allowed the identification of three types of structures: β-granules, γ-particles, and α-granules.[2]
β-Granules are composed of the polysaccharide glycogen, glycogenin, and γ-particles, which are protein-rich particles about 3 nm in diameter. β-Granules have a molecular mass of approximately 108 Da and a diameter of about 20–30 nm with a rosette-like appearance. They are considered a rapid energy source.[5][14]
Under physiological conditions, proteins account for 66–80% of their weight. These proteins also bind to one another, to the cytoskeleton, or to membranes, and all are involved in glycogen metabolism. Some of these include:
- glycogen synthase, the debranching enzyme, and glycogen phosphorylase;
- several regulatory proteins, such as:
- laforin and phosphoprotein phosphatase 1 (PP1; EC 3.1.3.17);
- phosphorylase kinase (EC 2.7.11.19) and AMPK (EC 2.7.11.31);
- the membrane anchoring protein STDB1 (note that phosphorylase kinase also binds to membranes);
- malin, or E3-ubiquitin ligase (EC 2.3.2.27), which binds glycogen via laforin and TRIM7.[30]
Unlike the pyruvate dehydrogenase complex or ribosomes, the stoichiometry and composition of β-granules are not constant but rather dynamic, as proteins associate with or dissociate from the granule depending on cellular conditions. In addition, differences are observed not only between different cell types but also within the same cell type, for example, in skeletal muscle cells depending on their subcellular localizations.[3]
α-Granules
In the liver, β-granules are organized into larger structures called α-granules.
They consist of several β-granules, have an approximate molecular mass of 108 kDa, and can reach up to about 300 nm in diameter, with a broccoli-like appearance. α-Granules are considered a slower energy source than β-granules.[31]
To date, the mechanism underlying their formation is not yet fully understood, although it appears that β-granules are linked through a protein scaffold rich in disulfide bonds.[3]
Glycogen metabolism: the synthetic phase
Glycogen is stored in cells during times of nutritional abundance. Its synthesis takes place in the cytosol, appears to be associated with actin filaments, and can occur from glucose derived from dietary carbohydrates or from glucose produced from noncarbohydrate precursors such as lactate and alanine.[3][32]
Lactate, produced by skeletal muscle cells under low oxygen conditions, by red blood cells that rely on anaerobic glycolysis for ATP production, and by other tissues, is mostly metabolized in the liver to form glucose via gluconeogenesis. Glucose diffuses from hepatocytes into the bloodstream and is transported to skeletal muscle cells, where it may be converted back to lactate, thus completing the Cori cycle.[33]
Alanine, a nonessential amino acid, may arise from transamination reactions in which pyruvate produced in glycolysis acts as the acceptor of the amino group from amino acids used for energy. Therefore, alanine serves as a carrier of both the carbon skeleton of pyruvic acid and amino groups from extrahepatic tissues to the liver, where the carbon skeleton is used to form glucose via gluconeogenesis and the amino groups are converted into urea through the urea cycle. Glucose then diffuses into the bloodstream and reaches peripheral tissues, where it may be converted back into pyruvate, thus completing the glucose-alanine cycle.[34]
Glucose enters cells through transmembrane carrier proteins known as glucose transporters (GLUTs), of which GLUT4 is primarily expressed in insulin-dependent or insulin-sensitive tissues such as skeletal and cardiac muscle, liver, and adipose tissue. GLUT4 is the insulin-responsive glucose transporter.[13]
Glucose uptake and phosphorylation
Once inside the cell, glucose is phosphorylated to glucose 6-phosphate (G6P). This reaction is catalyzed in hepatocytes and pancreatic β-cells by glucokinase (also known as hexokinase IV; EC 2.7.1.1) and by other hexokinases in other cell types.[35]
Fate of glucose 6-phosphate
The fate of glucose 6-phosphate depends on the metabolic status of the cell. It may enter the glycolytic pathway to provide energy and/or building blocks for other metabolic pathways. Alternatively, it can be isomerized to glucose 1-phosphate, a positional isomerization, through the reversible reaction catalyzed by phosphoglucomutase (EC 5.4.2.2). G1P can then be used for glycogen synthesis or can enter the pentose phosphate pathway when the reduced coenzyme NADPH is required for reductive biosynthesis (e.g., cholesterol and fatty acid synthesis) or when ribose 5-phosphate is needed for nucleotide synthesis.[9][36]
Glycogen synthesis
When glycogen synthesis predominates, glucose 1-phosphate is converted to UDP-glucose in the reaction catalyzed by UDP-glucose pyrophosphorylase (EC 2.7.7.9), at the expense of one UTP. UDP-glucose serves as the donor of glucose residues. Initially, glycogenin catalyzes the addition of glucose to the hydroxyl group of Tyr194, after which the enzyme adds 6–10 more glucose residues to form an oligosaccharide chain of 7–11 units.[37] This oligosaccharide chain then acts as the substrate for glycogen synthase, which, together with the branching enzyme, leads to glycogen synthesis.[8]
Glycogen metabolism: the degradative phase
Unlike glycogen synthesis, the catabolic or degradative phase of glycogen metabolism occurs both in the cytosol and in lysosomes, but through different metabolic pathways. Glycogenolysis is associated with the endoplasmic and sarcoplasmic reticulum and releases glucose for energy.[38][39]
Reactions catalyzed by glycogen phosphorylase, α-(1,4)-glucan-6-glycosyltransferase, and amylo-α-(1,6)-glucosidase (or debranching enzyme) lead to the release of glucose 1-phosphate, accounting for about 90% of the total, and free glucose, which represents the remaining 10%.[14]
In skeletal muscle cells, hexokinase activity is so high that any free glucose molecule is immediately phosphorylated to G6P, which cannot diffuse out of the cell. In hepatocytes, kidney cortex cells, and enterocytes, where the gluconeogenic pathway operates, glucose 6-phosphatase (EC 3.1.3.9) catalyzes the dephosphorylation of G6P (derived from the isomerization of G1P) to free glucose, which can then leave the cell and help regulate blood glucose levels.[40]
Lysosomal degradation
Although glycogen is synthesized in the cytosol, it is also found in lysosomes. Lysosomal glycogen may result from an autophagic mechanism and accounts for about 10% and 5% of the total glycogen content in the liver and muscle, respectively.[41]
According to one hypothesis, the degree of phosphorylation of glycogen is a key factor in regulating its lysosomal metabolism. This degree of phosphorylation appears to correlate with the age of the polysaccharide, acting as a quality control system: an increase in phosphorylation, which decreases the solubility of granules, is considered a marker that directs glycogen metabolism toward lysosomal degradation, a process also known as glycophagy.[5][42]
In lysosomes, glycogen breakdown is catalyzed by the enzyme acid α-(1,4)-glucosidase (EC 3.2.1.20). Hydrolysis by this enzyme leads to the release of D-glucose. Because the enzyme preferentially hydrolyzes α-(1,4) glycosidic bonds, it is not yet clear how α-(1,6) glycosidic bonds are cleaved.[43]
The importance of lysosomal glycogen catabolism is underscored by Pompe disease, or type II glycogenosis, which is caused by mutations in the acid α-glucosidase gene. A deficiency of functional acid α-(1,4)-glucosidase results in the excessive accumulation of glycogen in lysosomes and vesicular structures. In its most severe form, this glycogenosis is fatal within the first year of life.[7]
Coordinated regulation of glycogen metabolism
Glycogen synthesis and glycogenolysis are exergonic processes; if they operated simultaneously, they would lead to a waste of energy. In the cell, the two metabolic pathways are under stringent control and are reciprocally regulated so that when one is active, the other slows down. During evolution, this control has been achieved through the selection of different enzymes to catalyze the key steps of the two pathways, similar to the reciprocal regulation observed in glycolysis and gluconeogenesis.[13]
The key enzymes are glycogen phosphorylase and glycogen synthase, whose activity is regulated through:
- allosteric modifications, which occur on a time scale of milliseconds, are instantly reversible, and involve effectors such as calcium ions, glucose, and metabolites that signal the metabolic status of the cell, namely ATP, AMP, and G6P;
- covalent modifications, i.e., phosphorylation and dephosphorylation of specific target proteins, including the aforementioned enzymes, phosphorylase kinase, phosphoprotein phosphatase 1, and glycogen synthase kinase 3 (EC 2.7.11.26). Covalent regulation occurs on a time scale of seconds and is triggered by the binding of hormones, most importantly insulin, glucagon, and adrenaline (epinephrine), to their corresponding receptors on the plasma membrane.[14]
Allosteric and covalent mechanisms are superimposed in the coordinated regulation of glycogen metabolism.[15]
Covalent regulation
The metabolic changes induced by the binding of insulin, glucagon, and adrenaline to receptors on the plasma membrane of hepatocytes and skeletal muscle cells are discussed below.
Insulin
Insulin is secreted by pancreatic β-cells in response to high blood glucose levels, for example after a carbohydrate-rich meal, and has an anabolic effect. When insulin binds to receptors on the cell surface of hepatocytes and skeletal muscle cells, it triggers a cascade of reactions that results in the dephosphorylation of:
- glycogen phosphorylase, which is inhibited;
- glycogen synthase, which is activated.
In addition, insulin recruits GLUT4 to the plasma membrane of skeletal muscle cells. In this way, glycogen synthesis is stimulated and glycogenolysis is inhibited, helping to lower blood glucose levels.[44]
Glucagon
Glucagon is secreted by pancreatic α-cells in response to low blood glucose levels and has a catabolic effect. When glucagon binds to receptors on the cell surface of hepatocytes, it triggers a cascade of reactions that results in the phosphorylation of:
- glycogen phosphorylase, which is activated;
- glycogen synthase, which is inhibited.
As a result, glycogen synthesis is inhibited and glycogenolysis is stimulated, helping to raise blood glucose levels.[9]
Adrenaline
Adrenaline is secreted by the adrenal glands in response to stimulation of the sympathetic nervous system and plays a role in the fight-or-flight response. It may also be secreted during high-intensity exercise. Like glucagon, it has a catabolic effect, causing the phosphorylation of:
- glycogen phosphorylase, activating it;
- glycogen synthase, inhibiting it.
However, unlike glucagon, adrenaline acts not only on hepatocytes but also on skeletal muscle cells, binding to α1– and β2-adrenergic receptors in hepatocytes, and to β2-adrenergic receptors in skeletal muscle cells.[15]
Vasopressin
Vasopressin and adrenaline, when they binds to α1-adrenergic receptors, trigger intracellular pathways that lead to the release of calcium ions from the endoplasmic reticulum, thereby stimulating glycogenolysis and inhibiting glycogen synthesis.[8]
Allosteric regulation
The metabolic changes induced by the binding of allosteric effectors, AMP, ATP, glucose, G6P, and calcium ions, to the respective binding sites on target enzymes in skeletal muscle and liver are discussed below.
Skeletal muscle
In response to an increase in AMP concentration:
- glycogen phosphorylase b is activated.
As a result of an increase in ATP concentration:
- glycogen phosphorylase b is inhibited.
In response to an increase in glucose 6-phosphate concentration:
- PP1 is activated;
- glycogen phosphorylase b is inhibited;
- the phosphorylated form of glycogen synthase, glycogen synthase b, is activated allosterically via a conformational change that favors dephosphorylation by PP1. This activation allows glycogen synthase to act as a glucose 6-phosphate sensor.
Therefore, when cellular concentrations of ATP and G6P are low and AMP concentration is high, glycogen synthase is inhibited and glycogen phosphorylase is stimulated. Consequently, glycogenolysis is activated and glycogen synthesis is inhibited. Conversely, when ATP and glucose 6-phosphate concentrations are high, glycogen synthesis is stimulated and glycogenolysis is inhibited.[5]
In skeletal muscle cells, an increase in intracellular calcium ion concentration, released from the sarcoplasmic reticulum, triggers muscle contraction. Moreover, calcium ions bind to calmodulin, the δ subunit of muscle phosphorylase kinase. The calcium–calmodulin complex activates the kinase, which phosphorylates glycogen phosphorylase and glycogen synthase, activating the former and inhibiting the latter.[9]
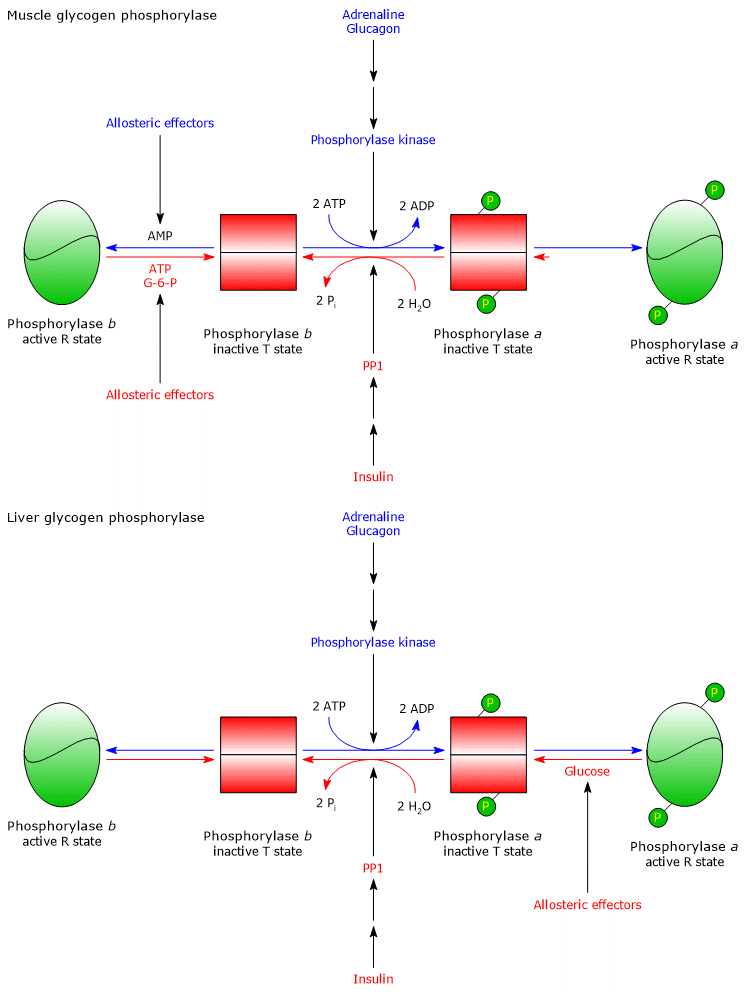
Liver
In the liver, glycogen phosphorylase b is not activated by AMP, whereas glycogen phosphorylase a is inhibited by an increase in blood glucose levels. Glucose concentration in the liver reflects blood glucose levels. In response to an increase in blood glucose, glucose binds to an inhibitory allosteric site on glycogen phosphorylase a, triggering a conformational change that exposes phosphorylated serine residues. These residues are then dephosphorylated by PP1, leading to glycogen phosphorylase inactivation. Therefore, glycogen phosphorylase acts as a blood glucose sensor, responding appropriately to changes in glucose levels.[9]
Localization of glycogen in humans
In humans, glycogen occurs in all cells, although the main stores are found in the liver and skeletal muscle, where it can represent up to 10% of liver mass and 2% of muscle mass, depending on nutritional status.[6] Therefore, skeletal muscle has a more limited capacity to store glycogen than the liver. However, because muscle mass is greater than liver mass, the total glycogen content in muscle is about double that of the liver. For example, in a non-fasting 70 kg adult male, there are approximately 100 g of glycogen in the liver and about 250 g in skeletal muscle.[45] Athletes can reach higher values, as in elite male marathon runners, whose liver and muscle stores can total about 475 g, corresponding to approximately 1,900 kcal.[46]
The amount of glycogen stored is much lower than that of fats because fats are a much more efficient form of energy storage. This is due to several factors.
- Fats can be stored in an anhydrous form, whereas the amount of water bound to glycogen is 2–3 times its weight.
- Stored fats are insoluble in water and thus osmotically inert.
- The oxidation of one gram of glycogen yields about 4 kcal, whereas one gram of fat yields about 9 kcal, roughly double the energy.[8]
Why it is important to humans
Fats, proteins and glycogen are energy stores that the body utilizes when needed.
In animals, fats are second only to proteins as energy reserves, although proteins represent an energy source of last resort, such as during prolonged fasting.
In a healthy adult, body fat accounts for about 21% of total body weight in men and 26% in women. This means that, in an adult male weighing 70 kg, body fat is sufficient to cover energy requirements for approximately two months. Conversely, glycogen stores are sufficient to meet energy demands for only about one day. Nevertheless, glycogen is still accumulated.[8]
Why?
- Unlike glucose, fatty acids, a class of lipids, cannot be metabolized anaerobically and therefore cannot supply energy to skeletal muscle during anaerobic exercise. It should also be noted that the energy yield from glycogen (or, more precisely, from glucose derived from glycogen) differs under aerobic and anaerobic conditions. Moreover, muscle cannot oxidize fatty acids as rapidly as it can oxidize glucose stored in glycogen.[14]
- Animals cannot convert fatty acids into glucose, and thus fats cannot be used to maintain glycemic homeostasis. Although glucose released from muscle glycogen remains within the cell, glucose released from hepatic glycogen, and to a lesser extent from renal glycogen, thanks to the enzyme glucose 6-phosphatase, enters the systemic circulation and helps regulate blood glucose levels.[40]
- Glycogen also plays a specialized role in fetal lung type II pulmonary cells (type II pneumocytes) which begin accumulating it around the 26th week of gestation. Here, glycogen serves as a major substrate for the synthesis of pulmonary surfactant lipids, chiefly dipalmitoylphosphatidylcholine.[47][48]
- The brain contains small glycogen stores, primarily in astrocytes. They accumulate during sleep and are mobilized upon waking, suggesting a role in brain activity and providing moderate protection against hypoglycemia.[49][50]
Glycogen and muscle work
Carbohydrates, namely glucose, and fatty acids are the main energy sources for muscle during exercise, and their relative contributions vary depending on the intensity and duration of the activity, as summarized below:
- <30% VO2max: mainly fatty acids;
- 40-60% VO2max: fatty acids and carbohydrates;
- 75% VO2max: mainly carbohydrates;
- >80% VO2max: almost exclusively carbohydrates.
Therefore, the contribution of glycogen to the energy required to support muscle work increases with exercise intensity, whereas the contribution of fatty acids decreases. Furthermore, when no carbohydrates are ingested, performance depends on glycogen stores in skeletal muscle and liver, whose relative utilization differs: as exercise intensity increases, muscle glycogen consumption rises, whereas liver glycogen consumption remains relatively constant.
Note: the relative contribution of fatty acids and glycogen as energy sources also varies according to the athlete’s level of training.[8][50]
Energy yield under anaerobic conditions
Under anaerobic conditions, the oxidation of glucose to lactate via anaerobic glycolysis yields two molecules of ATP.
Below is the ATP yield from the anaerobic oxidation of glucose released during glycogenolysis.
Glycogen phosphorylase and the oxidation of G1P under anaerobic conditions
Glycogen synthesis from glucose requires two ATP for each molecule of glucose.
The release of glucose 1-phosphate by the action of glycogen phosphorylase allows the recovery of one of the two ATP molecules used in the preparatory phase of glycolysis. The yield of anaerobic oxidation of glucose 6-phosphate, generated from G1P through the action of phosphoglucomutase, is therefore three ATP molecules, not two, because:
- only one molecule of ATP, instead of two, is consumed in the preparatory phase of glycolysis, as the hexokinase reaction is bypassed;
- four molecules of ATP are produced in the payoff phase of glycolysis.
Consequently, the cost-gain ratio is 1/3, meaning an energy yield of approximately 66.7%.
The reaction is:
Glycogen(n glucose residues) + 3 ADP + 3 Pi → Glycogen(n−1 glucose residues) + 2 Lactate + 3 ATP
Considering the two ATP molecules used in glycogen synthesis and the anaerobic oxidation of glucose 1-phosphate to lactate, the net yield is one molecule of ATP per molecule of glucose stored.
The resulting reaction is:[51]
Glucose + ADP + Pi → 2 Lactate + ATP
Debranching enzyme and the oxidation of glucose under anaerobic conditions
Considering the glucose released by the action of the debranching enzyme, the net ATP yield is zero because:
- two molecules of ATP are used in the synthesis of glycogen from glucose;
- the debranching enzyme releases free glucose, and thus two molecules of ATP are consumed in the preparatory phase of glycolysis;
- four molecules of ATP are produced in the payoff phase of glycolysis.
If we now consider the oxidation to lactate of all the glucose released from glycogen, the overall energy yield is:
1 − [(1/3) × 0.9] + [1 × 0.1] = 0.60
Therefore, under anaerobic conditions, the energy yield is approximately 60%, indicating that glycogen is an efficient storage form of energy.[51]
Energy yield under aerobic conditions
Under aerobic conditions, the oxidation of glucose to CO2 and H2O via glycolysis, pyruvate dehydrogenase complex, the Krebs cycle, the mitochondrial electron transport chain, and oxidative phosphorylation yields about 30 molecules of ATP.[9]
Below, the ATP yield from the aerobic oxidation of glucose released from glycogen by the action of glycogen phosphorylase and the debranching enzyme is considered.
Glycogen phosphorylase and the oxidation of G1P under aerobic conditions
Oxidation of glucose 6-phosphate, generated from glucose 1-phosphate by the action of phosphoglucomutase, to CO2 and H2O yields 31 molecules of ATP, not 30, because only one molecule of ATP is consumed in the preparatory phase of glycolysis. The cost-to-gain ratio is 1:31, corresponding to an energy efficiency of approximately 97%.
The overall reaction is:
Glycogen(n glucose residues) + 31 ADP + 31 Pi → Glycogen(n−1 glucose residues) + 31 ATP + 6 CO2 + 6 H2O
Considering the two ATP molecules used for glycogen synthesis and the aerobic oxidation of glucose 1-phosphate to CO2 and H2O, the net yield is 29 molecules of ATP per molecule of glucose stored.
The total reaction is:[51]
Glucose + 29 ADP + 30 Pi → 29 ATP + 6 CO2 + 6 H2O
Debranching enzyme and the oxidation of glucose under aerobic conditions
Considering the glucose released by the action of the debranching enzyme, the yield is 30 molecules of ATP, since two molecules of ATP are consumed in the preparatory phase of glycolysis. The cost-to-gain ratio is 2/30, corresponding to an energy yield of approximately 93.3%.
If we now consider the oxidation of all glucose released from glycogen to CO2 e H2O, the overall energy yield is:
1 − [1/31 × 0.9] + [2/30 × 0.1] = 0,96
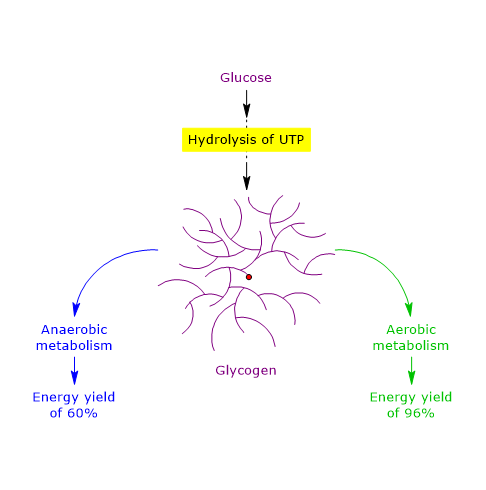 Therefore, under aerobic conditions, the energy yield is 96%, indicating that glycogen is an extremely efficient storage form of energy, with a 36% higher yield compared to anaerobic conditions.[51]
Therefore, under aerobic conditions, the energy yield is 96%, indicating that glycogen is an extremely efficient storage form of energy, with a 36% higher yield compared to anaerobic conditions.[51]
References
- ^ a b Daghlas S.A., Mohiuddin S.S. Biochemistry, Glycogen. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539802/
- ^ a b c d e f Roach P.J., Depaoli-Roach A.A., Hurley T.D, Tagliabracci V.C. Glycogen and its metabolism: some new developments and old themes. Biochem J 2012;441:763-87. doi:10.1042/BJ20111416
- ^ a b c d e f g Neoh G.K.S., Tan X., Chen S., Roura E., Dong X., Gilbert R.G. Glycogen metabolism and structure: a review. Carbohydr Polym 2024;346:122631. doi:10.1016/j.carbpol.2024.122631
- ^ Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley J. & Sons, Inc. 2012
- ^ a b c d e f Prats C., Graham T.E., and Shearer J. The dynamic life of the glycogen granule. J Biol Chem 2018;293(19):7089-98. doi:10.1074/jbc.R117.802843
- ^ a b c Adeva-Andany M.M., González-Lucán M., Donapetry-García C., Fernández-Fernández C., Ameneiros-Rodríguez E. Glycogen metabolism in humans. BBA Clin 2016;27;5:85-100. doi:10.1016/j.bbacli.2016.02.001
- ^ a b Canibano-Fraile R., Harlaar L., Dos Santos C.A., et al. Lysosomal glycogen accumulation in Pompe disease results in disturbed cytoplasmic glycogen metabolism. J Inherit Metab Dis 2023;46(1):101-115. doi:10.1002/jimd.12560
- ^ a b c d e f Stipanuk M.H., Caudill M.A. Biochemical, physiological, and molecular aspects of human nutrition. 4th Edition. St. Louis: Elsevier, 2018.
- ^ a b c d e f g h i Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
- ^ Terlouw E.M.C., Picard B., Deiss V., Berri C., Hocquette J.F., Lebret B., Lefèvre F., Hamill R., Gagaoua M. Understanding the determination of meat quality using biochemical characteristics of the muscle: stress at slaughter and other missing keys. Foods 2021;10(1):84. doi:10.3390/foods10010084
- ^ Claude Bernard, the founder of modern medicine. Cells 2022;11(10):1702. doi:10.3390/cells11101702
- ^ The Nobel Foundation. Retrieved October 30, 2025. https://www.nobelprize.org/the-nobel-prize-organisation/the-nobel-foundation/
- ^ a b c d Berg J.M., Tymoczko J.L., Gregory J.G. Jr, Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
- ^ a b c d e f g Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
- ^ a b c Garrett R.H., Grisham C.M. Biochemistry. 6th Edition. Brooks/Cole, Cengage Learning, 2016.
- ^ Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. 7th Edition. Garland Science, Taylor & Francis Group, 2022.
- ^ Yu Y., Delbianco M. Conformational studies of oligosaccharides. Chemistry 2020;26(44):9814-9825. doi:10.1002/chem.202001370
- ^ a b Gunja-Smith Z., Marshall J.J., Mercier C., Smith E.E., Whelan W.J. A revision of the Meyer-Bernfeld model of glycogen and amylopectin. FEBS Lett 1970:12(2);101-104. doi:10.1016/0014-5793(70)80573-7
- ^ Whelan W.J. Enzymic explorations of the structures of starch and glycogen. Biochem J 1971;122(5):609-622. doi:10.1042/bj1220609
- ^ Meléndez-Hevia E., Waddell T.G., Shelton E.D. Optimization of molecular design in the evolution of metabolism: the glycogen molecule. Biochem J 1993;295(2):477-83. doi:10.1042/bj2950477
- ^ Shearer J., Graham T.E. Novel aspects of skeletal muscle glycogen and its regulation during rest and exercise. Exerc Sport Sci Rev 2004;32(3):120-6. doi:10.1097/00003677-200407000-00008
- ^ Marchand I., Chorneyko K., Tarnopolsky M., Hamilton S., Shearer J., Potvin J., Graham T.E. Quantification of subcellular glycogen in resting human muscle: granule size, number, and location. J Appl Physiol (1985) 2002;93(5):1598-607. doi:10.1152/japplphysiol.00585.2001
- ^ Meléndez R., Meléndez-Hevia E., Mas F., Mach J., Cascante M. Physical constraints in the synthesis of glycogen that influence its structural homogeneity: a two-dimensional approach. Biophys J 1998;75(1):106-14. doi:10.1016/S0006-3495(98)77498-3
- ^ Melendez R., Melendez-Hevia E., Cascante M. How did glycogen structure evolve to satisfy the requirement for rapid mobilization of glucose? A problem of physical constraints in structure building. J Mol Evol 1997;45(4):446-55. doi:10.1007/PL00006249
- ^ Fastman N.M., Liu Y., Ramanan V., Merritt H., et al. The structural mechanism of human glycogen synthesis by the GYS1-GYG1 complex. Cell Rep 2022;40(1):111041. doi:10.1016/j.celrep.2022.111041
- ^ Fontana J.D. The presence of phosphate in glycogen. FEBS Lett 1980;1:109(1):85-92. doi:10.1016/0014-5793(80)81317-2
- ^ Gentry M.S., Guinovart J.J., Minassian B.A., Roach P.J., Serratosa J.M. Lafora disease offers a unique window into neuronal glycogen metabolism. J Biol Chem 2018;293(19):7117-25. doi:10.1074/jbc.R117.803064
- ^ Turnbull J., Tiberia E., Pereira S., et al. Deficiency of a glycogen synthase-associated protein, Epm2aip1, causes decreased glycogen synthesis and hepatic insulin resistance. J Biol Chem 2013;288(48):34627-37. doi:10.1074/jbc.M113.483198
- ^ Roach P.J. Glycogen phosphorylation and Lafora disease. Mol Aspects Med 2015;46:78-84. doi:10.1016/j.mam.2015.08.003
- ^ Brewer M.K., Gentry M.S. Brain glycogen structure and its associated proteins: past, present and future. Adv Neurobiol 2019;23:17-81. doi:10.1007/978-3-030-27480-1_2
- ^ Sullivan M.A., Aroney S.T., Li S., Warren FJ, et al. Changes in glycogen structure over feeding cycle sheds new light on blood-glucose control. Biomacromolecules 2014;15(2):660-5. doi:10.1021/bm401714v
- ^ Baqué S., Guinovart J.J., Ferrer J.C. Glycogenin, the primer of glycogen synthesis, binds to actin. FEBS Lett 1997;417(3):355-9. doi:10.1016/s0014-5793(97)01299-4
- ^ Katz J., Tayek J.A. Gluconeogenesis and the Cori cycle in 12-, 20-, and 40-h-fasted humans. Am J Physiol 1998;275(3):E537-42. doi:10.1152/ajpendo.1998.275.3.E537
- ^ Felig P., Pozefsk T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
- ^ Wilson J.E. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol 2003;206(Pt 12):2049-57. doi:10.1242/jeb.00241
- ^ Horecker B.L. The pentose phosphate pathway. J Biol Chem 2002;277(50):47965-47971. doi:10.1074/jbc.X200007200
- ^ Smythe C., Cohen P. The discovery of glycogenin and the priming mechanism for glycogen biogenesis. Eur J Biochem 1991;200(3):625-31. doi:10.1111/j.1432-1033.1991.tb16225.x
- ^ Zois C.E., Harris A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J Mol Med (Berl) 2016;94(2):137-54. doi:10.1007/s00109-015-1377-9
- ^ Mandl J. Glycogen-endoplasmic reticulum connection in the liver. Int J Mol Sci 2023;24(2):1074. doi:10.3390/ijms24021074
- ^ a b Rajas F., Gautier-Stein A., Mithieux G. Glucose 6-phosphate, a central hub for liver carbohydrate metabolism. Metabolites 2019;9(12):282. doi:10.3390/metabo9120282
- ^ Zirin J., Nieuwenhuis J., Perrimon N. Role of autophagy in glycogen breakdown and its relevance to chloroquine myopathy. PLoS Biol 2013;11(11):e1001708. doi:10.1371/journal.pbio.1001708
- ^ Koutsifeli P., Varma U., Daniels L.J., et al. Glycogen-autophagy: molecular machinery and cellular mechanisms of glycophagy. J Biol Chem 2022;298(7):102093. doi:10.1016/j.jbc.2022.102093
- ^ Okuyama M., Saburi W., Mori H., Kimura A. α-Glucosidases and α-1,4-glucan lyases: structures, functions, and physiological actions. Cell Mol Life Sci 2016;73(14):2727-51. doi:10.1007/s00018-016-2247-5
- ^ Rahman M.S., Hossain K.S., Das S., et al. Role of insulin in health and disease: an update. Int J Mol Sci 2021;22(12):6403. doi:10.3390/ijms22126403
- ^ Knuiman P., Hopman M.T., Mensink M. Glycogen availability and skeletal muscle adaptations with endurance and resistance exercise. Nutr Metab (Lond) 2015;12:59. doi:10.1186/s12986-015-0055-9
- ^ Beelen M., Burke L.M., Gibala M.J., van Loon J.C. Nutritional strategies to promote postexercise recovery. Int J Sport Nutr Exerc Metab 2010:20(6);515-32. doi:10.1123/ijsnem.20.6.515
- ^ Farrell P.M., Bourbon J.R. Fetal lung surfactant lipid synthesis from glycogen during organ culture. Biochim Biophys Acta 1986;878(2):159-67. doi:10.1016/0005-2760(86)90142-6
- ^ Yildiz Atar H., Baatz J.E., Ryan R.M. Molecular mechanisms of maternal diabetes effects on fetal and neonatal surfactant. Children (Basel) 2021;8(4):281. doi:10.3390/children8040281
- ^ Rosenthal M.D., Glew R.H. Medical Biochemistry − Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
- ^ Duran J., Gruart A., López-Ramos J.C., Delgado-García J.M., Guinovart J.J. Glycogen in astrocytes and neurons: physiological and pathological aspects. Adv Neurobiol 2019;23:311-329. doi:10.1007/978-3-030-27480-1_10
- ^ a b c d Snider M.D., Croniger C.M. Devlin′s textbook of biochemistry with clinical correlations. 8th Edition. John Wiley & Sons, Inc. 2024.