The Leloir pathway is the primary route for galactose metabolism.
Discovered by Luis F. Leloir and colleagues in 1948, this pathway converts galactose into glucose 1-phosphate. Specifically, it involves the inversion of the configuration of the hydroxyl group at carbon 4 of galactose, one of the monosaccharide’s chiral centers.[1]
The metabolic intermediates involved in this isomerization serve as building blocks in various biosynthetic pathways, such as the glycosylation of proteins and lipids or glycogen synthesis. Their role depends on the developmental stage, the cell’s metabolic state, and the tissue type.[2]
With the exception of the first step, the reactions in the Leloir pathway are reversible, proceeding in either direction depending on substrate concentrations and the tissue’s metabolic needs. This enables the interconversion of galactose and glucose.[3]
The importance of the Leloir pathway, and of galactose itself, is highlighted by its high degree of evolutionary conservation, from bacteria to plants and animals.[4] In humans, its relevance is further underscored by the severe consequences of mutations in the genes encoding its enzymes, which lead to the inherited metabolic disorder known as galactosemia.[5]
Summary: Key Points
- Primary function: The Leloir pathway is the essential metabolic route that converts dietary galactose into glucose 1-phosphate, making it available for energy production or glycogen storage.
- Cellular trapping and irreversibility: The galactokinase reaction is the only irreversible step; it adds a negative charge to galactose, effectively preventing it from diffusing out of the cell.
- Biochemical flexibility: Except for the second step, the reactions are fully reversible, enabling cells to interconvert sugar nucleotides based on developmental stages and specific tissue needs.
- Clinical relevance: Inherited mutations in the pathway’s genes cause galactosemia. The resulting accumulation of galactitol triggers osmotic and oxidative stress, leading to early cataract formation.
Contents
- Galactose
- The steps of the Leloir pathway
- Role of the Leloir pathway
- Leloir pathway and galactosemia
- References
Galactose
Galactose, along with glucose and fructose, is one of the monosaccharides that can be absorbed in the intestine. The main dietary source of galactose is lactose, which, together with maltose, trehalose and sucrose, is one of the disaccharides commonly found in food.
Since there are no transporters for disaccharides, their glycosidic bonds are hydrolyzed in the final stage of carbohydrate digestion, releasing their constituent monosaccharides. In the case of lactose, these are glucose and galactose.[6]
The monosaccharides are then absorbed and transported via the portal circulation to the liver, the primary site of galactose metabolism. The liver absorbs most of the galactose (approximately 88%) through facilitated passive diffusion mediated by the GLUT2 transporter.[7]
The small remaining amount of circulating galactose reaches other organs and tissues, such as the mammary gland, which during lactation uses galactose for lactose synthesis and for the glycosylation of milk proteins and lipids.[4]
The steps of the Leloir pathway
The Leloir pathway consists of four enzymatic reactions, catalyzed respectively by:
- galactose mutarotase (also known as aldose 1-epimerase; EC 5.1.3.3);
- galactokinase (EC 2.7.1.6);
- galactose 1-phosphate uridylyltransferase (EC 2.7.7.12);
- UDP-galactose 4-epimerase (EC 5.1.3.2).[8]
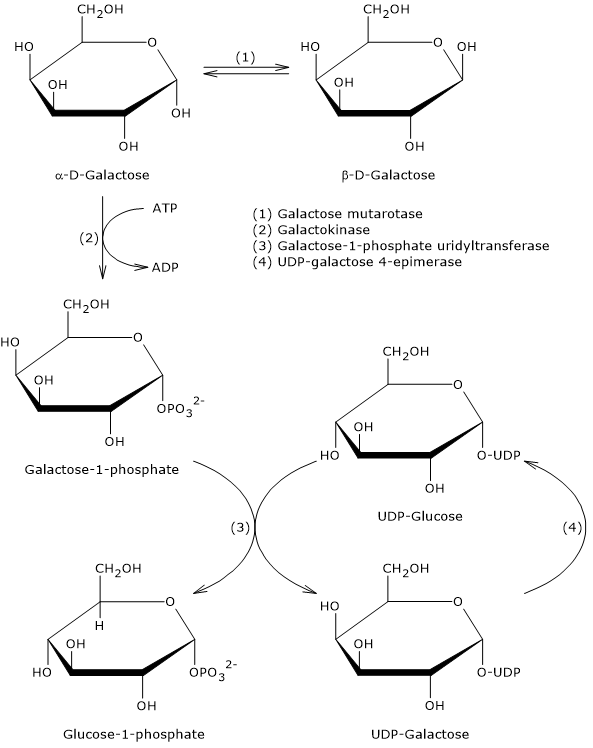
Step 1: galactose mutarotase
The hydrolysis of the β-(1→4) glycosidic bond in lactose results in the release of glucose and β-D-galactose.
Galactokinase, the enzyme responsible for the second step of the Leloir pathway, is specific for the α-anomer of galactose.[8] Therefore, the β-anomer must first be converted into the α-anomer. This conversion is catalyzed by galactose mutarotase.
In addition to galactose, this enzyme is also capable of interconverting the α- and β-anomeric forms of other sugars, including glucose, xylulose, maltose, and lactose, although with varying degrees of efficiency.[9]
Step 2: galactokinase
In the second step, α-D-galactose is phosphorylated to galactose 1-phosphate in a reaction catalyzed by galactokinase.[8] Phosphorylation of galactose, like that of glucose to glucose 6-phosphate, is metabolically important for several reasons.
- Galactose 1-phosphate cannot diffuse out of the cell because it carries a negative charge, and there are no transporters for phosphorylated sugars in the plasma membrane.[10]
- Phosphorylation increases the energy content of the monosaccharide, initiating destabilization and facilitating its subsequent metabolism.[11]
- By converting galactose into galactose 1-phosphate, the reaction lowers the intracellular concentration of free galactose, promoting its continued uptake via facilitated transport.[10]
The reaction catalyzed by galactokinase represents the irreversible step of the Leloir pathway.[3] Unlike hexokinase and glucokinase (EC 2.7.1.1), which phosphorylate the hydroxyl group at carbon 6 of glucose, galactokinase and fructokinase (EC 2.7.1.4) phosphorylate the hydroxyl group at carbon 1 of galactose and fructose, respectively.[12]
The conversion of galactose 1-phosphate to glucose-1-phosphate requires two additional reactions: the third and fourth steps of the Leloir pathway.
Step 3: galactose 1-phosphate uridylyltransferase
In the third step, galactose 1-phosphate uridylyltransferase catalyzes the transfer of a uridine monophosphate (UMP) group from UDP-glucose to galactose 1-phosphate, yielding UDP-galactose and glucose 1-phosphate.
The reaction follows a ping-pong (double displacement) mechanism, involving the formation of a covalent intermediate between the enzyme and the UMP group.[10]
Step 4: UDP-galactose 4-epimerase
In the final step, UDP-galactose is converted into UDP-glucose in a reaction catalyzed by UDP-galactose 4-epimerase. This enzyme inverts the configuration of the hydroxyl group at carbon 4 and is responsible for the interconversion between UDP-galactose and UDP-glucose, and, by extension, between galactose and glucose.[13]
UDP-galactose 4-epimerase requires NAD+ as a cofactor, and the reaction proceeds through the formation of a ketonic intermediate at C4, accompanied by the reduction of NAD+ to NADH. The sugar then rotates within the active site, exposing the opposite face to NADH, which transfers a hydride ion back to carbon 4, but in the reversed configuration.[14]
Because NAD+ is first reduced and then re-oxidized, there is no net redox change for the coenzyme, which therefore does not appear in the overall reaction equation.
UDP-galactose 4-epimerase is thought to be the rate-limiting enzyme of the Leloir pathway.[4]
The UDP-glucose produced in this reaction is subsequently recycled by UDP-glucose pyrophosphorylase, releasing glucose 1-phosphate.[2]
In mammals, UDP-galactose 4-epimerase also catalyzes the interconversion between UDP-N-acetylgalactosamine and UDP-N-acetylglucosamine.[15]
It is important to note that UDP-galactose and UDP-glucose, like galactose and glucose in general, have the same molecular formula. They differ only in the configuration of the hydroxyl group at carbon 4, making them stereoisomers, and more specifically, epimers, a form of optical isomerism.
Role of the Leloir pathway
The Leloir pathway enables cells to utilize galactose or its derivative, glucose, in various metabolic pathways, both anabolic and catabolic, depending on the metabolic state of the cell or tissue.
Because the reaction catalyzed by UDP-galactose 4-epimerase is reversible, the conversion of glucose into galactose (and its nucleotide derivatives) is also possible.[2]
UDP-galactose can be used in:
- the glycosylation of proteins and lipids, such as the formation of galactocerebrosides, which are major glycolipid components of myelin. For this reason, galactose was initially referred to as cerebrose;[4][16]
- the lactating mammary gland, where it is used for lactose synthesis in a reaction catalyzed by the lactose synthase system.[17]
Furthermore, since the UDP-galactose 4-epimerase reaction is reversible, glucose, once activated to UDP-glucose by UDP-glucose pyrophosphorylase (EC 2.7.7.9), can be converted to UDP-galactose for lactose production.[18]
In the liver and skeletal muscle, UDP-glucose derived from UDP-galactose can be used:[3]
- for protein and lipid glycosylation;
- in glycogen synthesis, especially when the cell’s energy demand is low. Compared to glucose and fructose, galactose is preferentially incorporated into hepatic glycogen rather than entering oxidative pathways;
- after conversion to glucose 1-phosphate (via UDP-glucose pyrophosphorylase), and subsequent isomerization to glucose 6-phosphate (via phosphoglucomutase, EC 5.4.2.2), it may enter various metabolic routes, including glycolysis, the pentose phosphate pathway or gluconeogenesis.[4]
Note: UDP-galactose, discovered during studies on the Leloir pathway, was the first nucleotide sugar to be identified.[19]
Leloir pathway and galactosemia
Glycosylations are post-translational modifications that play a crucial role in enabling and regulating a wide range of biological processes. Defects in glycosylation have been associated with numerous pathological conditions, including cancer, diabetes, and congenital metabolic disorders, particularly the congenital disorders of glycosylation (CDGs), which are primarily autosomal recessive monogenic conditions.[20] Among these, galactosemia is a well-known disorder, first described by von Reuss A. in 1908.[21] Galactosemia results from mutations in one of the genes encoding the enzymes of the Leloir pathway, and four clinical types have been identified.
- Type I: due to galactose-1-phosphate uridylyltransferase deficiency. This is the most common and severe form and is referred to as classic galactosemia.[22]
- Type II: due to galactokinase deficiency.[23]
- Type III: due to UDP-galactose 4-epimerase deficiency.[24]
- Type IV: due to galactose mutarotase deficiency.[25]
At present, the standard treatment for all forms of galactosemia consists of a galactose-restricted diet, aimed at preventing the toxic accumulation of galactose or its metabolites.[26]
Galactosemia and cataracts
The accumulation of galactose activates alternative metabolic pathways, including the synthesis of galactitol and galactonate.
One of the early clinical manifestations of galactosemia is the development of cataracts, typically within the first two years of life. In the most severe cases, the disease may also lead to brain, liver, and kidney damage.[9]
One proposed mechanism underlying cataract formation involves the reduction of galactose, accumulated in the lens, to galactitol in a reaction catalyzed by aldose reductase (EC 1.1.1.21).[27] Galactitol is poorly metabolized and does not readily diffuse across cell membranes due to its low lipophilicity. As an osmotically active compound, it increases intracellular osmotic pressure, leading to a net influx of water into the lens cells.[28]
Moreover, its synthesis consumes NADPH, potentially depleting cellular NADPH pools and impairing the activity of glutathione reductase (EC 1.8.1.7). This reduction in antioxidant capacity may contribute to the accumulation of free radicals.[29]
The combined effects of osmotic stress and oxidative damage can ultimately compromise cell integrity, resulting in cell death and the formation of lens opacities. Additionally, galactitol has been reported to act as an inhibitor of galactose mutarotase, which may exacerbate galactose accumulation by further hindering its metabolism.[30]
References
- ^ Leloir L.F., de Fekete M.A., Cardini C.E. Starch and oligosaccharide synthesis from uridine diphosphate glucose. J Biol Chem 1961;236:636-41. doi:10.1016/S0021-9258(18)64280-2
- ^ a b c Frey P.A. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J 1996;10(4):461-70. doi:10.1096/fasebj.10.4.8647345
- ^ a b c Conte F., van Buuringen N., Voermans N.C., Lefeber D.J. Galactose in human metabolism, glycosylation and congenital metabolic diseases: time for a closer look. Biochim Biophys Acta Gen Subj 2021;1865(8):129898. doi:10.1016/j.bbagen.2021.129898
- ^ a b c d e Coelho A.I., Berry G.T., Rubio-Gozalbo M.E. Galactose metabolism and health. Curr Opin Clin Nutr Metab Care 2015;18(4):422-427. doi:10.1097/MCO.0000000000000189
- ^ Petry K.G., Reichardt J.K. The fundamental importance of human galactose metabolism: lessons from genetics and biochemistry. Trends Genet 1998;14(3):98-102. doi:10.1016/s0168-9525(97)01379-6
- ^ Coelho A.I., Rubio-Gozalbo M.E., Vicente J.B., Rivera I. Sweet and sour: an update on classic galactosemia. J Inherit Metab Dis 2017;40(3):325-342. doi:10.1007/s10545-017-0029-3
- ^ Drozdowski L.A., Thomson A.B. Intestinal sugar transport. World J Gastroenterol 2006;12(11):1657-70. doi:10.3748/wjg.v12.i11.1657
- ^ a b c Holden H.M., Rayment I., Thoden J.B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem 2003;278(45):43885-43888. doi:10.1074/jbc.R300025200
- ^ a b Thoden J.B., Timson D.J., Reece R.J., and M. Holden H.M. Molecular structure of human galactose mutarotase. J Biol Chem 2004;279(22):23431-23437. doi:10.1074/jbc.M402347200
- ^ a b c Garrett R.H., Grisham C.M. Biochemistry. 7th Edition. Cengage Learning, 2023.
- ^ Berg J.M., Tymoczko J.L., Gatto J.G., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
- ^ Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
- ^ Thoden J.B., Holden H.M. Dramatic differences in the binding of UDP-galactose and UDP-glucose to UDP-galactose 4-epimerase from Escherichia coli. Biochemistry 1998;37(33):11469-77. doi:10.1021/bi9808969
- ^ Thoden J.B., Wohlers T.M., Fridovich-Keil J.L., Holden H.M. Human UDP-galactose 4-epimerase. Accommodation of UDP-N-acetylglucosamine within the active site. J Biol Chem 2001;276(18):15131-6. doi:10.1074/jbc.M100220200
- ^ Demirbas D., Coelho A.I., Rubio-Gozalbo M.E., Berry G.T. Hereditary galactosemia. Metabolism. 2018;83:188-196. doi:10.1016/j.metabol.2018.01.025
- ^ Helenius A., Aebi M. Intracellular functions of N-linked glycans. Science 2001;291(5512):2364-9. doi:10.1126/science.291.5512.2364
- ^ Lin Y., Sun X., Hou X., Qu B., Gao X., Li Q. Effects of glucose on lactose synthesis in mammary epithelial cells from dairy cow. BMC Vet Res 2016;12:81. doi:10.1186/s12917-016-0704-x
- ^ Sunehag A., Tigas S., Haymond M.W. Contribution of plasma galactose and glucose to milk lactose synthesis during galactose ingestion. J Clin Endocrinol Metab 2003;88(1):225-9. doi:10.1210/jc.2002-020768
- ^ Cardini C.E., Paladini A.C., Caputto R., Leloir L.F. Uridine diphosphate glucose: the coenzyme of the galactose-glucose phosphate isomerization. Nature 1950;165:191-192. doi:10.1038/165191a0
- ^ Dall’Olio F. Glycobiology of aging. Subcell Biochem 2018;90:505-526. doi:10.1007/978-981-13-2835-0_17
- ^ Timson D.J. Type IV galactosemia. Genet Med 2019;21:1283-1285. doi:10.1038/s41436-018-0359-z
- ^ Los E., Ford G.A. Galactose-1-phosphate uridyltransferase deficiency. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441957/
- ^ Ramani P.K., Arya K. Galactokinase deficiency. [Updated 2023 Jul 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560683/
- ^ Fridovich-Keil J., Bean L., He M., et al. Epimerase deficiency galactosemia. 2011 Jan 25 [Updated 2021 Mar 4]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK51671/
- ^ Wada Y., Kikuchi A., Arai-Ichinoi N., Sakamoto O., Takezawa Y., Iwasawa S., Niihori T., Nyuzuki H., Nakajima Y., Ogawa E., Ishige M., Hirai H., Sasai H., Fujiki R., Shirota M., Funayama R., Yamamoto M., Ito T., Ohara O., Nakayama K., Aoki Y., Koshiba S., Fukao T., Kure S. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet Med 2019;21(6):1286-1294. doi:10.1038/s41436-018-0340-x
- ^ Succoio M., Sacchettini R., Rossi A., Parenti G., Ruoppolo M. Galactosemia: biochemistry, molecular genetics, newborn screening, and treatment. Biomolecules 2022;12(7):968. doi:10.3390/biom12070968
- ^ Pintor J. Sugars, the crystalline lens and the development of cataracts. Biochem Pharmacol 2012;1:4. doi:10.4172/2167-0501.1000e119
- ^ Tsakiris S., Marinou K., Schulpis K. H. The in vitro effects of galactose and its derivatives on rat brain Mg2+-ATPase activity. Pharmacol Toxicol 2022;91:254-257. doi:10.1034/j.1600-0773.2002.910506.x
- ^ Lai K., Elsas L.J., Wierenga K.J. Galactose toxicity in animals. IUBMB Life 2009;61(11):1063-74. doi:10.1002/iub.262
- ^ Keston A.S. Inhibition of action of lens mutarotase on glucose by cataractogenic sugars and corresponding polyols. Arch Biochem Biophys 1963;102(2):306-312. doi:10.1016/0003-9861(63)90184-X
Domande Frequenti
What is the Leloir pathway and what is its primary function?
The Leloir pathway is the primary metabolic route responsible for converting galactose into glucose 1-phosphate. This process enables cells to utilize dietary galactose to generate energy via glycolysis or store it in the liver and skeletal muscle as glycogen.
Which step in the Leloir pathway is considered irreversible?
The only irreversible reaction in the Leloir pathway is the second step, catalyzed by the enzyme galactokinase (EC 2.7.1.6). This reaction phosphorylates α-D-galactose into galactose 1-phosphate, preventing the sugar from diffusing out of the cell.
What are the four types of galactosemia and their causes?
Galactosemia is an inherited disorder classified into four types, each caused by a specific enzyme deficiency in the Leloir pathway: Type I (classic) is due to GALT deficiency; Type II to GALK deficiency; Type III to GALE deficiency; and Type IV to GALM (galactose mutarotase) deficiency.
How does untreated galactosemia lead to cataract formation?
Accumulated galactose is converted into galactitol by the enzyme aldose reductase. Galactitol cannot cross cell membranes, increasing osmotic pressure inside lens cells. This leads to a massive influx of water, causing osmotic stress, oxidative damage, and eventual lens opacity (cataracts).