The pentose phosphate pathway, also known as the phosphogluconate pathway, is a metabolic route common to all living organisms that oxidizes glucose and serves as an alternative to glycolysis. It branches off after the formation of glucose 6-phosphate (G6P) and is also referred to as the hexose monophosphate shunt, where shunt denotes a metabolic diversion or bypass.
Its primary function is the production, in varying proportions, of NADPH, a reduced coenzyme, and ribose 5-phosphate, a five-carbon phosphorylated sugar (i.e., a pentose phosphate), from which the pathway derives its name.[1]
Conceptually, the pathway consists of two phases: an oxidative phase, in which NADPH is generated, and a non-oxidative phase, which produces ribose 5-phosphate and other phosphorylated carbohydrates.[2]
It is estimated that over 10% of cellular glucose is metabolized via this pathway, which notably oxidizes glucose without directly producing or consuming ATP.[3]
In Summary: Key Points
- Definition: a cytosolic pathway for the oxidative metabolism of glucose, alternative to glycolysis, whose main function is the production, in variable proportions, of NADPH and ribose 5-phosphate.
- Fate of NADPH: produced in the oxidative phase, it provides the reducing power required for reductive biosyntheses and antioxidant defense mechanisms.
- Fate of ribose 5-phosphate: produced in the non-oxidative phase, it is used for the synthesis of nucleotides and various coenzymes.
- Key enzymes: glucose 6-phosphate dehydrogenase, regulated by the NADP+/NADPH ratio, and transketolase, a thiamine pyrophosphate–dependent enzyme.
- Metabolic flexibility: the pathway adapts to cellular needs, operating in different modes to favor the production of NADPH, ribose 5-phosphate, or the recovery of carbon in the form of glycolytic intermediates.
Contents
- Historical background
- Main function
- Sites of the pentose phosphate pathway
- Oxidative phase of the pentose phosphate pathway
- Non-oxidative phase of the pentose phosphate pathway
- The cell’s need for NADPH, ribose 5-phosphate, and ATP
- References
Historical background
The first evidence of the pentose phosphate pathway emerged in the 1930s through the work of Otto Warburg, who received the Nobel Prize in Physiology or Medicine in 1931 for his work on cellular metabolism and respiratory enzymes.[4]
Warburg and his collaborator Erwin Christian discovered NADP during studies on the oxidation of glucose 6-phosphate to 6-phosphogluconate.
Further evidence came from observations that glucose metabolism continued in tissues even in the presence of glycolysis inhibitors such as fluoride and iodoacetate ions, which inhibit enolase (EC 4.2.1.11) and glyceraldehyde 3-phosphate dehydrogenase (EC 1.2.1.12), respectively.[5]
However, the pathway was fully elucidated only in the 1950s, thanks to the work of several researchers, notably Efraim Racker, Bernard Horecker, Frank Dickens, and Fritz Lipmann, who received the Nobel Prize in Physiology or Medicine in 1953 for his discovery of coenzyme A, not specifically for the pentose phosphate pathway, although his work was highly relevant in the broader context of intermediary metabolism.[6][7]
Main function
The primary function of the pentose phosphate pathway is the production of NADPH and ribose 5-phosphate.[8]
NADPH is needed for reductive biosynthesis, such as the synthesis of fatty acids, cholesterol, steroid hormones and two non-essential amino acids, proline and tyrosine, from glutamate and phenylalanine, respectively, as well as for the reduction of oxidized glutathione. In these reactions, the reduced coenzyme acts as an electron donor, more precisely as a hydride ion donor (:H−), a species composed of one proton and two electrons.[1][9]
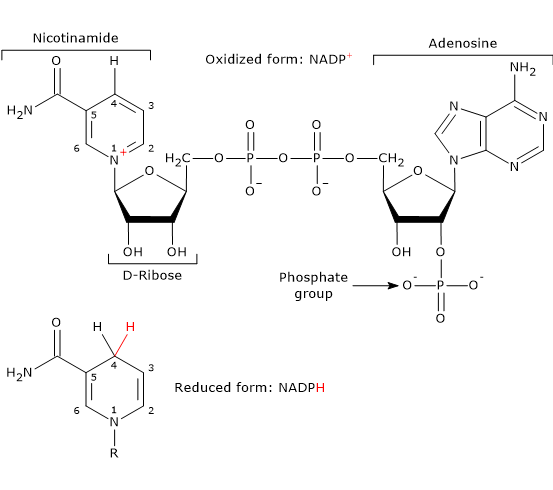
Note: In vertebrates, approximately half of the NADPH required for the reductive steps of fatty acid synthesis is produced via the pentose phosphate pathway. The remainder is generated by the reaction catalyzed by the malic enzyme (EC 1.1.1.40).[10]
Malate + NADP+ ⇌ Pyruvate + NADPH + CO2
Ribose 5-phosphate is used in the synthesis of nucleotides and nucleic acids (DNA and RNA), as well as ATP, and coenzymes such as coenzyme A, NAD, NADP, and FAD. It also plays a role in the biosynthesis of the essential amino acids tryptophan and histidine.[8] This five-carbon phosphorylated sugar is not used directly; instead, it is converted to 5-phosphoribosyl 1-pyrophosphate (PRPP) in a reaction catalyzed by ribose phosphate pyrophosphokinase, also known as PRPP synthase (EC 2.7.6.1).[10]
Ribose 5-phosphate + ATP → 5-Phosphoribosyl 1-pyrophosphate + AMP
Additional functions
In addition to producing NADPH and ribose 5-phosphate, the pentose phosphate pathway also serves other functions, both anabolic and catabolic.
In yeasts and many bacteria, it is involved in the catabolism of the five-carbon sugars ribose, xylose, and arabinose.[9]
In humans, it also contributes to the catabolism of the aforementioned pentoses, as well as of less common sugars containing three, four, or seven carbon atoms, derived from the diet or from endogenous sources. These include:
- pentoses released during the breakdown of structural carbohydrates;
- ribose 5-phosphate generated from nucleotide catabolism.[11][12]
In photosynthetic organisms, it contributes to carbon dioxide (CO2) fixation during the Calvin cycle.[9]
In addition to ribose 5-phosphate, the pathway also provides several intermediates for other biosynthetic processes, including:
- erythrose 4-phosphate, used in the synthesis of the aromatic amino acids phenylalanine, tryptophan, and tyrosine;
- ribulose 5-phosphate, a precursor in riboflavin (vitamin B2) biosynthesis;
- sedoheptulose 7-phosphate, which, in Gram-negative bacteria, is involved in the synthesis of heptose units found in the lipopolysaccharide layer of the outer membrane.[1][13]
Sites of the pentose phosphate pathway
In animal cells and bacteria, the hexose monophosphate shunt, like glycolysis, fatty acid synthesis, and most reactions of gluconeogenesis, occurs in the cytosol. Notably, glycolysis, gluconeogenesis, and the pentose phosphate pathway are interconnected through several shared enzymes and intermediates.[12]
In plant cells, the pentose phosphate pathway takes place in the plastids, and its intermediates can reach the cytosol through membrane pores in these organelles.[10]
In humans, the expression of the enzymes involved in the pathway varies significantly between tissues. Relatively high levels are found in the liver, adrenal cortex, testes, ovaries, thyroid, mammary glands during lactation, and in red blood cells.[2] In all these tissues, a constant supply of NADPH is required to sustain reductive biosynthesis and/or to counteract reactive oxygen species (ROS), which can damage sensitive cellular structures such as DNA, membrane lipids, and proteins. This protection is largely achieved through the reduction of oxidized glutathione (GSSG) to reduced glutathione (GSH), the major intracellular antioxidant in erythrocytes and most other cells. This reaction is catalyzed by glutathione reductase (EC 1.8.1.7).[14]
High levels of phosphogluconate pathway enzymes are also found in rapidly dividing cells, such as enterocytes, skin cells, bone marrow cells, embryonic cells, and, under pathological conditions, cancer cells. These cells require a continuous supply of ribose 5-phosphate for nucleic acid synthesis.
Conversely, these enzymes are present at very low levels in skeletal muscle, where the pentose phosphate pathway is virtually absent. In this tissue, glucose 6-phosphate is primarily utilized for energy production through glycolysis and the citric acid cycle.[2]
Oxidative phase of the pentose phosphate pathway
The oxidative phase of the pentose phosphate pathway consists of two irreversible oxidation reactions, the first and third steps, and one hydrolysis.
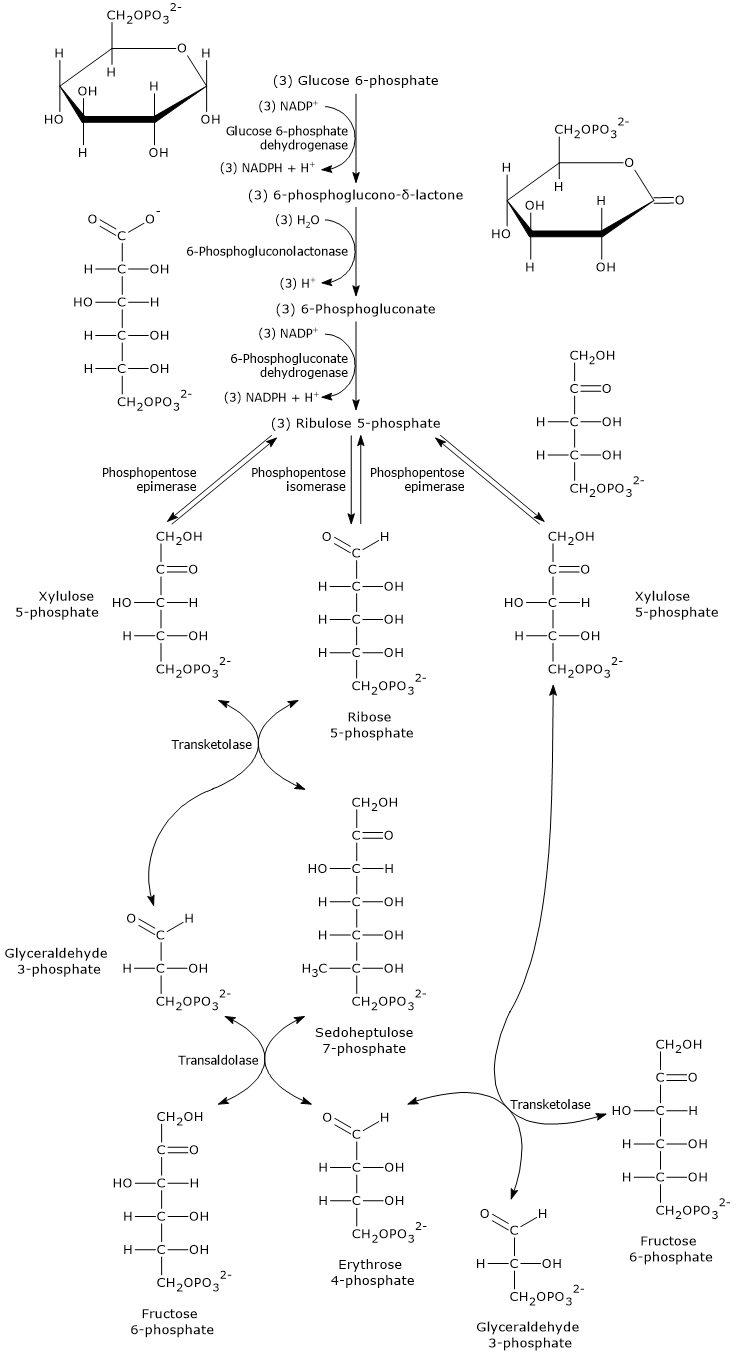
During this phase, glucose 6-phosphate is converted into ribulose 5-phosphate, a five-carbon phosphorylated sugar that serves as the starting substrate for the reactions of the non-oxidative phase.
This conversion is accompanied by the formation of two molecules of NADPH and the release of carbon 1 of glucose as CO2.[15]
The overall equation is:
3 Glucose 6-phosphate + 6 NADP+ + 3 H2O → 6 NADPH + 6 H+ + 3 CO2 + 3 Ribulose 5-phosphate
Oxidation of glucose 6-phosphate to 6-phosphoglucono-δ-lactone
In the first step of the oxidative phase, glucose 6-phosphate dehydrogenase (G6PD; EC 1.1.1.49) catalyzes the oxidation of glucose 6-phosphate to 6-phosphoglucono-δ-lactone, an intramolecular ester.
This occurs via the transfer of a hydride ion (H−) from carbon 1 of glucose 6-phosphate to NADP+, which acts as the oxidizing agent.[12]
Glucose 6-phosphate + NADP+ → 6-Phosphoglucono-δ-lactone + NADPH + H+
Note: this reaction produces the first molecule of NADPH in the pentose phosphate pathway.
The reaction catalyzed by G6PD is unique to the pathway. As in most metabolic pathways, the first reaction that is unique, known as the committed step, is essentially irreversible.
In the liver, it has a ΔG of −17.6 kJ/mol (−4.21 kcal/mol), and it is highly allosterically regulated. Indeed, glucose 6-phosphate dehydrogenase serves as the major control point for the flux of metabolites through the pentose phosphate pathway.[5]
G6PD expression and immune function
In humans, the highest levels of G6PD are found in neutrophils and macrophages, two types of phagocytic cells.
During inflammation, these cells use NADPH to generate superoxide radicals (O2•−) from molecular oxygen, in a reaction catalyzed by NADPH oxidase (EC 1.6.3.1).
2 O2 + NADPH → 2 O2•− + NADP+ + H+
Superoxide radicals are used to generate, for antimicrobial defense, both other reactive oxygen species (ROS) and reactive nitrogen species (RNS), such as:[2]
- hydrogen peroxide (H2O2), via the reaction catalyzed by superoxide dismutase (SOD; EC 1.15.1.1)
2 O2•− + 2 H+ → H2O2 + O2
- peroxynitrite (ONOO−), via the reaction with nitric oxide (•NO)
O2•− + •NO → ONOO−
- hydroperoxide radical (HOO•)
O2•− + H+ → HOO•
Catalytic mechanism of G6PD
The catalytic mechanism of glucose 6-phosphate dehydrogenase has been studied in great detail in the microorganism Leuconostoc mesenteroides, whose enzyme displays the unusual ability to use NAD+ and/or NADP+ as coenzyme.
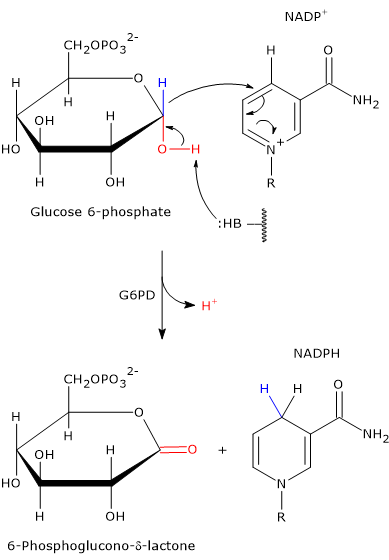
The enzyme does not require metal ions for its activity. Instead, an amino acid residue in the active site acts as a general base, capable of abstracting a proton from the hydroxyl group attached to carbon 1 of glucose 6-phosphate.[16]
In the bacterial enzyme, this function is performed by the Nɛ2 atom of the imidazole ring in a histidine side chain. This nitrogen atom contains a lone pair of electrons that enables it to act as a nucleophile.
Its action facilitates the oxidation of glucose 6-phosphate, a cyclic hemiacetal with C-1 in the aldehyde oxidation state, into a cyclic ester, specifically a lactone.
Simultaneously, the hydride ion is transferred from C-1 of glucose 6-phosphate to the C-4 position of the nicotinamide ring of NADP+ to form NADPH.
Since this histidine residue is conserved in many of the glucose 6-phosphate dehydrogenase sequences identified so far, it is likely that this catalytic mechanism is common to G6PD enzymes across a wide range of organisms.[17]
Regulation of G6PD
Glucose 6-phosphate dehydrogenase is the primary control point in the pentose phosphate pathway and the main regulator of NADPH production.[18]
In humans, G6PD exists in two forms:
- an inactive monomeric form;
- an active form, which exists in a dimer–tetramer equilibrium.[14]
One of the main modulators of its activity is the cytosolic NADP+/NADPH ratio.
NADPH acts as a competitive inhibitor, while NADP+ is both a substrate and a structural activator, stabilizing the enzyme’s active conformation by binding to a regulatory site near the dimer interface, helping to maintain the dimeric (active) structure.
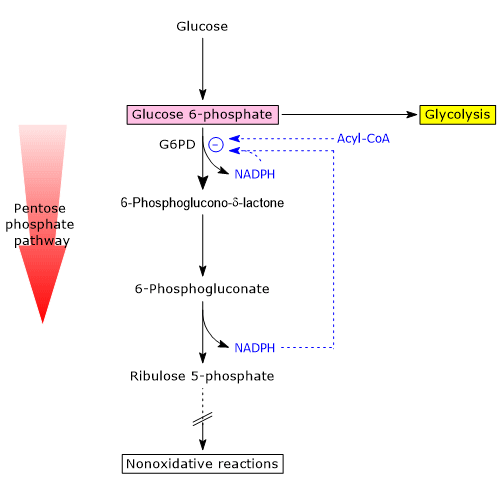
Under typical metabolic conditions, the NADP+/NADPH ratio is low.
As a result, NADP+ availability is limited, less coenzyme binds to the enzyme, and G6PD activity is reduced, regardless of expression levels. In these conditions, the oxidative phase of the pathway is virtually inactive.
Conversely, in cells where NADPH-consuming reactions are active (e.g., fatty acid synthesis, antioxidant defense), the cytosolic concentration of NADPH decreases, while NADP+ increases.
This change promotes the activation of G6PD, triggering the oxidative phase of the hexose monophosphate pathway.
Therefore, the fate of glucose 6-phosphate is influenced by the cell’s current demand for NADPH.[19][20]
A second regulatory mechanism involves the accumulation of acyl-CoAs, intermediates in fatty acid biosynthesis.[8]
These molecules bind to the dimeric form of G6PD and induce its dissociation into monomers, resulting in the loss of catalytic activity.[14]
Insulin also plays a role by upregulating the expression of the genes encoding glucose 6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase.
Thus, in the well-fed state, insulin enhances the carbon flux through the pentose phosphate pathway, increasing NADPH production
Note: insulin also promotes fatty acid synthesis, which depends on a continuous supply of NADPH.[15]
Hydrolysis of 6-phosphoglucono-δ-lactone to 6-phosphogluconate
In the second step of the oxidative phase, 6-phosphoglucono-δ-lactone is hydrolyzed to form 6-phosphogluconate, a linear molecule.
Although 6-phosphoglucono-δ-lactone is hydrolytically unstable and can undergo spontaneous (nonenzymatic) ring-opening at a significant rate, this hydrolysis reaction is greatly accelerated in the cell by the enzyme 6-phosphogluconolactonase (EC 3.1.1.31).[21]
6-Phosphoglucono-δ-lactone + H2O → 6-Phosphogluconate
Oxidative decarboxylation of 6-phosphogluconate to ribulose 5-phosphate
In the final step of the oxidative phase, 6-phosphogluconate undergoes an oxidative decarboxylation, producing:
- ribulose 5-phosphate, a keto-pentose;
- CO2;
- the second molecule of NADPH in the pathway.[19]
The reaction is catalyzed by 6-phosphogluconate dehydrogenase (EC 1.1.1.44), an enzyme that requires Mg2+ ions for activity.[5]
6-Phosphogluconate + NADP+ → Ribulose 5-phosphate + NADPH + CO2
Catalytic mechanism of 6-phosphogluconate dehydrogenase
The catalytic mechanism of 6-phosphogluconate dehydrogenase closely resembles that of isocitrate dehydrogenase (EC 1.1.1.41), a key enzyme of the citric acid cycle.[5]
It proceeds via acid-base catalysis through a three-step mechanism, involving two strictly conserved amino acid residues, a lysine (Lys) and a glutamate (Glu).[22]
In humans:
- Lys185, which acts as both a general base and a general acid;
- Glu192, which acts as a general acid.[23]
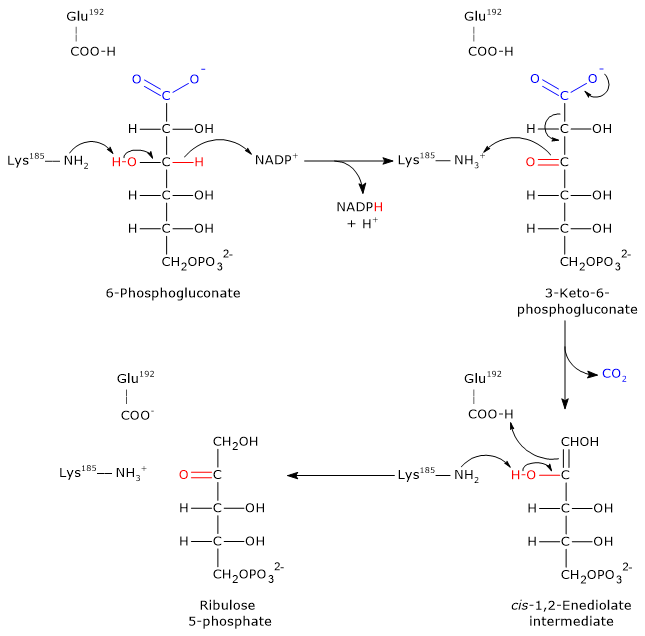
Step 1: oxidation
In the first step, 6-phosphogluconate is oxidized to a β-keto acid, specifically 3-keto-6-phosphogluconate.
- The ε-amino group of Lys185 acts as a general base, abstracting a proton from the hydroxyl group at C-3 of 6-phosphogluconate.
- This promotes the transfer of a hydride ion (H−) from C-3 of the substrate to C-4 of the nicotinamide ring in NADP+, yielding NADPH.
- The 3-keto intermediate and NADPH are released from the active site.
Step 2: decarboxylation
The 3-keto-6-phosphogluconate formed is highly unstable and undergoes a spontaneous decarboxylation, resulting in:
- the formation of CO2 (from C-1 of the original glucose 6-phosphate);
- the intermediate cis-1,2-enediol of ribulose 5-phosphate, a high-energy tautomer.
In this step Lys185 acts as a general acid, donating a proton to the C-3 carbonyl oxygen, facilitating the decarboxylation.
Step 3: Keto–enol tautomerization
In the final step, the cis-1,2-enediol intermediate undergoes a stereospecific keto–enol tautomerization to form ribulose 5-phosphate.
- Glu192 donates a proton to the C-1 of the enediol.
- Simultaneously, the ε-amino group of Lys185 acts as a base, abstracting a proton from the hydroxyl group on C-2.
The result is the formation of ribulose 5-phosphate, completing the oxidative phase of the pentose phosphate pathway.[22][23]
Non-oxidative phase of the pentose phosphate pathway
In the non-oxidative phase of the pentose phosphate pathway, several phosphorylated carbohydrates are produced, and their fate depends on the cell’s relative needs for NADPH, ribose 5-phosphate, and ATP.
This phase consists of five freely reversible steps, involving a series of interconversions of phosphorylated sugars.
It begins with two initial reactions: an isomerization and an epimerization.
These transform ribulose 5-phosphate into:
- ribose 5-phosphate, and
- xylulose 5-phosphate, respectively.[15]
Epimerases (EC 5.1), a subclass of isomerases, catalyze configurational changes at a single asymmetric carbon atom, typically via a deprotonation/protonation mechanism.
In contrast, isomerases mediate the rearrangement of chemical groups between different carbon atoms.[24]
Note: enzymatic isomerizations and epimerizations play a key role in carbohydrate metabolism, enabling the interconversion of sugars with distinct functional properties.[25]
Isomerization of ribulose 5-phosphate to ribose 5-phosphate
In the first step of the non-oxidative phase, ribulose 5-phosphate (a ketose) is converted into its corresponding aldose, ribose 5-phosphate, via an isomerization reaction.
The reaction is catalyzed by phosphopentose isomerase, also known as ribose 5-phosphate isomerase (EC 5.3.1.6).
Ribulose 5-phosphate ⇄ Ribose 5-phosphate
This is an example of functional group isomerism, where a ketone is converted into an aldehyde.[1]
Catalytic mechanism of phosphopentose isomerase
The catalytic mechanism of phosphopentose isomerase resembles that of phosphohexose isomerase (EC 5.3.1.9), a glycolytic enzyme. Both involve the formation of a high-energy intermediate, cis-1,2-enediol, through a proton-transfer mechanism that is common in aldose–ketose isomerizations.[26]
In Escherichia coli, the mechanism (described here in the direction of ribulose 5-phosphate formation from ribose 5-phosphate, as occurs in the Calvin cycle of photosynthesis) proceeds as follows.
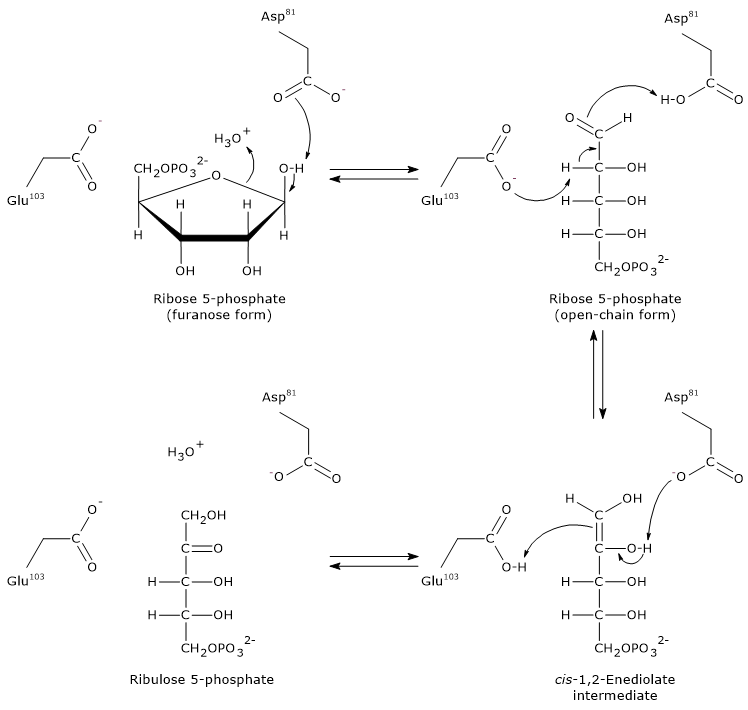
Step 1: ring opening
The furanose ring of ribose 5-phosphate is opened by interaction with an aspartic acid residue (Asp81), which accepts a proton from the hydroxyl group bound to C-1.
It is likely that a water molecule donates the proton required for this step.
Note: in aqueous solution, spontaneous ring opening of ribose is rare, less than 0.5% occurs naturally.
Step 2: formation of the enediol intermediate
A glutamic acid residue (Glu103) acts as a general base, abstracting a proton from the C-2 carbon. Simultaneously, Asp81 donates a proton. This concerted action leads to the formation of the cis-1,2-enediol intermediate.
Step 3: protonation and product formation
The now protonated Glu103 acts as a general acid, donating a proton to C-1 of the enediol intermediate. At the same time, Asp81 accepts a proton from the hydroxyl group on C-2. These proton transfers lead to the formation of ribulose 5-phosphate.
During ribose 5-phosphate synthesis from ribulose 5-phosphate, as occurs in the pentose phosphate pathway, phosphopentose isomerase functions in reverse, following the same general principles of acid-base catalysis.[27]
Epimerization of ribulose 5-phosphate to xylulose 5-phosphate
The other metabolic fate of ribulose 5-phosphate in the pentose phosphate pathway is its epimerization to xylulose 5-phosphate, a ketose like ribulose 5-phosphate.
This reaction is catalyzed by phosphopentose epimerase (EC 5.1.3.1).[5]
Ribulose 5-Phosphate ⇄ Xylulose 5-Phosphate
Note: xylulose 5-phosphate also acts as a regulatory molecule. It inhibits gluconeogenesis and stimulates glycolysis by modulating the intracellular concentration of fructose 2,6-bisphosphate in the liver.[28]
Catalytic mechanism of phosphopentose epimerase
Like the reactions catalyzed by 6-phosphogluconate dehydrogenase and ribose 5-phosphate isomerase, this reaction also proceeds through the formation of an enediol intermediate.
However, in this case, the double bond is formed between C-2 and C-3, rather than between C-1 and C-2.[5]
During the reaction, an amino acid residue in the enzyme’s active site acts as a general base and abstracts a proton from C-3, leading to the formation of the cis-2,3-enediol intermediate.
Next, an acidic amino acid residue donates a proton back to C-3, but from the opposite face, resulting in inversion at C-3 and formation of xylulose 5-phosphate.[29]
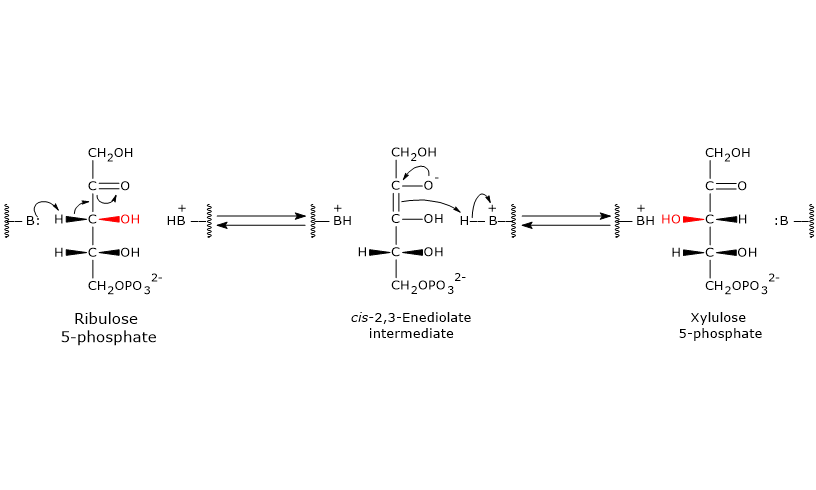
Up to this point, the hexose monophosphate shunt has generated the following, for each molecule of glucose 6-phosphate metabolized:
- a pool of three pentose 5-phosphates, ribulose 5-phosphate, ribose 5-phosphate, and xylulose 5-phosphate, which coexist at equilibrium;
- two molecules of NADPH.
In the next three steps (steps 6 through 8), the enzymes transketolase (EC 2.2.1.1) and transaldolase (EC 2.2.1.2), key enzymes of the pentose phosphate pathway, catalyze a series of carbon skeleton rearrangements.
These reactions lead to the formation of three-, four-, six-, and seven-carbon sugar phosphates, which can be redirected into other metabolic pathways, depending on the cell’s metabolic needs.[12]
By analyzing the flow of metabolites across different pathways, it becomes clear that the concerted action of transketolase and transaldolase allows the non-oxidative phase of the pentose phosphate pathway to interact with:
- glycolysis;
- gluconeogenesis;
- pathways for the synthesis of various vitamins, coenzymes, and nucleic acid precursors.[1]
Transketolase
Transketolase is the rate-limiting enzyme of the non-oxidative phase of the pentose phosphate pathway and is the first enzyme to act downstream of ribose 5-phosphate isomerase and phosphopentose epimerase.[30]
Discovered independently in 1953 by Horecker and Racker, and named by Racker, transketolase catalyzes the transfer of a two-carbon unit from a ketose donor to an aldose acceptor. This occurs in both the sixth and eighth steps of the pathway.
The ketose donors are typically:
- xylulose 5-phosphate;
- sedoheptulose 7-phosphate;
- fructose 6-phosphate.
The aldose acceptors include:
- ribose 5-phosphate;
- glyceraldehyde 3-phosphate;
- erythrose 4-phosphate.[7]
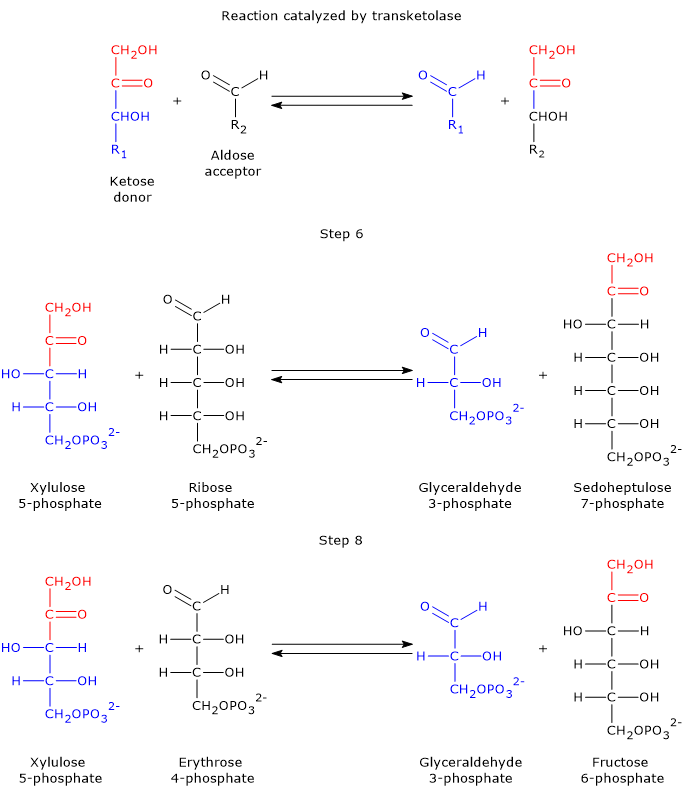
Taking the forward reactions as an example, in the sixth step, the ketose donor xylulose 5-phosphate transfers a two-carbon unit to the aldose acceptor ribose 5-phosphate. This reaction generates glyceraldehyde 3-phosphate, which corresponds to the three-carbon remainder of xylulose 5-phosphate, and sedoheptulose 7-phosphate, a seven-carbon sugar that participates in the subsequent step of the pathway.[31]
In the eighth step, xylulose 5-phosphate once again acts as the donor, but this time the acceptor is erythrose 4-phosphate. The products of this reaction are another molecule of glyceraldehyde 3-phosphate and fructose 6-phosphate.
It is worth noting that three out of the four products generated by transketolase, namely, two molecules of glyceraldehyde 3-phosphate and one of fructose 6-phosphate, are also key intermediates of glycolysis.[30]
Beyond xylulose 5-phosphate, sedoheptulose 7-phosphate, and fructose 6-phosphate, transketolase is also capable of using other 2-keto sugars and a variety of aldose phosphates as substrates.[32]
Transketolase requires thiamine pyrophosphate (TPP) as a cofactor. TPP, the biologically active form of thiamine (vitamin B1), is tightly bound to the enzyme and participates in the transfer of activated aldehyde intermediates by stabilizing the two-carbon carbanions formed during the reaction.[8]
Catalytic mechanism of transketolase
The carbon atom located between the sulfur and nitrogen atoms of the thiazolium ring of thiamine pyrophosphate, namely, the C-2 atom, is significantly more acidic than most =CH groups in other molecules. This is due to the adjacent positively charged nitrogen, which electrostatically stabilizes the carbanion formed upon proton dissociation. As a result, the C-2 proton is readily abstracted, generating a carbanion. This proton abstraction is catalyzed by transketolase.
The resulting carbanion attacks the carbonyl carbon of the donor ketose substrate. In step 6, this is xylulose 5-phosphate (or sedoheptulose 7-phosphate in the reverse reaction); in step 8, it is again xylulose 5-phosphate (or fructose 6-phosphate in reverse). Taking the forward direction of step 6 as an example, the covalent adduct formed between thiamine pyrophosphate and xylulose 5-phosphate undergoes cleavage at the C-2–C-3 bond. This reaction produces glyceraldehyde 3-phosphate, which is released, and a two-carbon hydroxyethyl group, which remains covalently bound to the C-2 atom of the thiazolium ring.
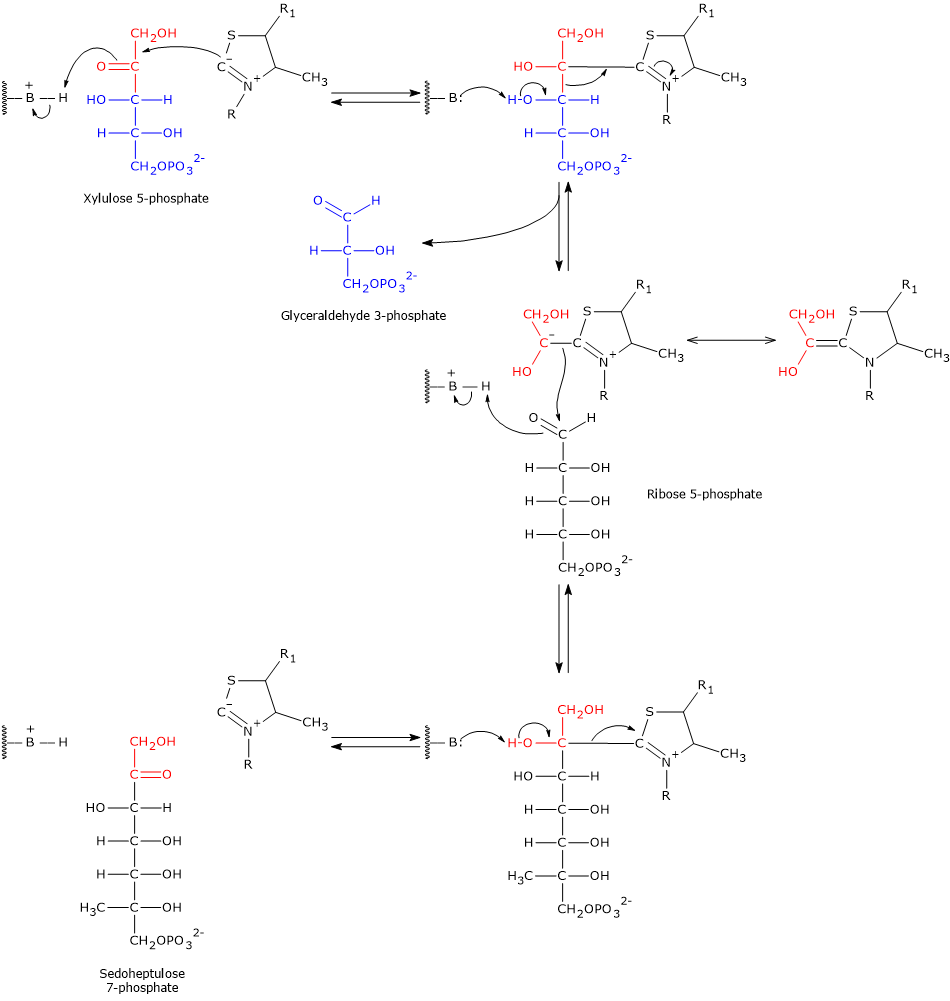
The negative charge on this hydroxyethyl intermediate is stabilized by the thiazolium ring of thiamine pyrophosphate. The positively charged nitrogen in the ring acts as an electron sink, delocalizing the carbanion’s negative charge via resonance. In this way, the thiazolium ring provides an electron-deficient (electrophilic) environment that stabilizes the intermediate.
Subsequently, the hydroxyethyl-TPP group condenses with ribose 5-phosphate, the aldehyde acceptor substrate, via nucleophilic attack on the aldehyde carbon. This forms a new covalent adduct with thiamine pyrophosphate.
Finally, cleavage of the adduct releases sedoheptulose 7-phosphate and regenerates the TPP carbanion.[8][12][30]
Transaldolase
Transaldolase was discovered in 1953 by Horecker and Smyrniotis in brewer’s yeast, identified as Saccharomyces cerevisiae. In the seventh step of the pentose phosphate pathway, this enzyme catalyzes the transfer of a three-carbon unit from the donor substrate sedoheptulose 7-phosphate to the acceptor substrate glyceraldehyde 3-phosphate. The reaction yields fructose 6-phosphate and erythrose 4-phosphate.
Sedoheptulose 7-phosphate + Glyceraldehyde 3-phosphate ⇄ Fructose 6-phosphate + Erythrose 4-phosphate
As in the reactions catalyzed by transketolase, the donor of the carbon unit is a ketose, while the acceptor is an aldose. In the reverse direction, the donor becomes fructose 6-phosphate and the acceptor erythrose 4-phosphate.[9]
Catalytic mechanism of transaldolase
Unlike transketolase, transaldolase does not require a cofactor for its activity.
The reaction proceeds in two steps: an aldol cleavage followed by an aldol condensation. Below, the catalytic mechanism of E. coli transaldolase B is described, taking as an example the forward reaction that produces erythrose 4-phosphate and fructose 6-phosphate.
Step 1: aldol cleavage and intermediate formation
In the first step, the ε-amino group of a lysine residue (Lys132) in the active site performs a nucleophilic attack on the carbonyl carbon (C-2) of sedoheptulose 7-phosphate. This occurs after a proton transfer to a glutamic acid residue (Glu96), mediated by a water molecule. The result is the formation of a carbinolamine intermediate.
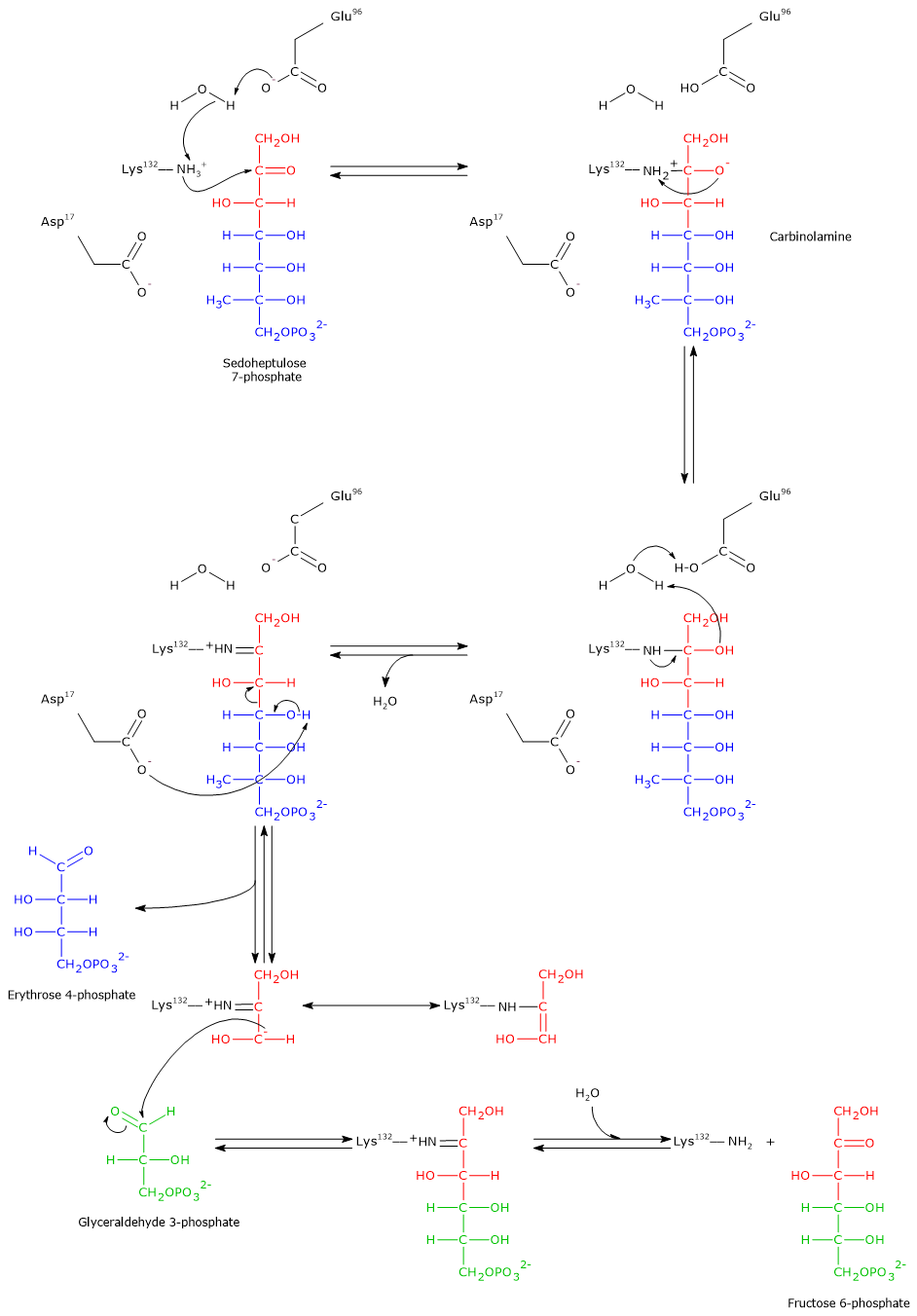
Subsequently, the elimination of a water molecule leads to the formation of an enzyme-bound imine, also known as a Schiff base. This step also involves the transfer of a proton from Glu96 to the catalytic water molecule.
Note: this enzyme–substrate covalent intermediate is similar to that formed during the reaction catalyzed by aldolase (EC 4.1.2.13) in glycolysis.
Then, an aspartic acid residue (Asp17) acts as a base and abstracts a proton from the hydroxyl group at C-4. This facilitates the cleavage of the C–C bond between C-3 and C-4, releasing erythrose 4-phosphate, an aldose. The remaining three-carbon carbanion stays covalently bound to the enzyme and is stabilized by resonance. Just like in the transketolase mechanism, the positive charge on the Schiff base nitrogen acts as an electron sink, stabilizing the negative charge on the carbanion.[5][8][12][33]
Step 2: aldol condensation and product release
After the release of erythrose 4-phosphate, the acceptor substrate glyceraldehyde 3-phosphate enters the active site.
The previously formed carbanion performs a nucleophilic attack on the carbonyl carbon of glyceraldehyde 3-phosphate. This aldol condensation results in the formation of a new carbon–carbon bond and a covalently bound ketose intermediate.
The final step involves the hydrolysis of the Schiff base. This reaction restores the free ε-amino group of Lys132 and releases fructose 6-phosphate, a ketose and the second product of the reaction. The enzyme is now regenerated and ready for a new catalytic cycle.
As previously mentioned, in the eighth step of the pentose phosphate pathway, transketolase catalyzes the synthesis of fructose 6-phosphate and glyceraldehyde 3-phosphate from erythrose 4-phosphate and xylulose 5-phosphate, linking the activities of transaldolase and transketolase in the non-oxidative phase of the pathway.[33]
The cell’s need for NADPH, ribose 5-phosphate, and ATP
From a molecular point of view, the fate of glucose 6-phosphate depends largely on the relative activities of the enzymes that metabolize it through glycolysis and the pentose phosphate pathway, particularly phosphofructokinase-1 (PFK-1; EC 2.7.1.11) and glucose 6-phosphate dehydrogenase, both of which are highly regulated.[1]
PFK-1 is inhibited when ATP and/or citrate concentrations increase, that is, when the cell’s energy charge is high. Conversely, it is activated when AMP and/or fructose 2,6-bisphosphate concentrations increase, indicating a low energy charge. As a result, when the energy charge is high, the carbon flux, specifically the flux of glucose 6-phosphate, through glycolysis decreases.[34][35]
G6PD, in contrast, is inhibited by NADPH and by acyl-CoAs, which are intermediates in fatty acid biosynthesis. Thus, when cytosolic NADPH levels rise, the pentose phosphate pathway is downregulated. However, when NADPH levels fall, the inhibition is relieved, the pathway is reactivated, and both NADPH and ribose 5-phosphate are synthesized.[8][19]
Therefore, depending on the cell’s current demand for ATP, NADPH, and ribose 5-phosphate, reactions from glycolysis and the pentose phosphate pathway can be strategically combined to prioritize the synthesis of specific metabolites. This metabolic flexibility is also made possible by the fact that the non-oxidative phase of the pentose phosphate pathway is primarily controlled by substrate availability.
The four main scenarios are outlined below.[12]
When the need for NADPH is much greater than that for ribose 5-phosphate or ATP
When the cellular requirement for NADPH far exceeds that for ribose 5-phosphate or ATP, namely, when no additional ATP is needed and the energy charge is high, glucose 6-phosphate enters the pentose phosphate pathway and is fully oxidized to CO2. These metabolic conditions are typically found in adipose tissue during fatty acid synthesis.
Through a combination of reactions from the non-oxidative phase and selected gluconeogenic reactions, namely, those catalyzed by triose phosphate isomerase (EC 5.3.1.1), aldolase (EC 4.1.2.13), phosphohexose isomerase (EC 5.3.1.9), and fructose 1,6-bisphosphatase (EC 3.1.3.11), six molecules of ribulose 5-phosphate can be converted into five molecules of glucose 6-phosphate. In this way, the non-oxidative phase supports the continuation of the oxidative phase by replenishing the starting substrate.
The process can be divided into three groups of reactions.
- Oxidative phase reactions
These reactions are catalyzed by the enzymes of the oxidative phase, producing two molecules of NADPH and one molecule of ribulose 5-phosphate per glucose 6-phosphate.
6 Glucose 6-phosphate + 12 NADP+ + 6 H2O → 6 Ribulose 5-phosphate + 6 CO2 + 12 NADPH + 12 H+
- Non-oxidative phase reactions
These include the enzymes phosphopentose epimerase, ribose 5-phosphate isomerase, transketolase, and transaldolase, which convert ribulose 5-phosphate into fructose 6-phosphate and glyceraldehyde 3-phosphate.
6 Ribulose 5-phosphate → 4 Fructose 6-phosphate + 2 Glyceraldehyde 3-phosphate
- Gluconeogenic reactions
The fructose 6-phosphate and glyceraldehyde 3-phosphate produced in the non-oxidative phase are recycled into glucose 6-phosphate via gluconeogenic reactions. These reactions allow the cycle to restart.
4 Fructose 6-phosphate + 2 Glyceraldehyde 3-phosphate + H2O → 5 Glucose 6-phosphate + Pi
The net result of the last two groups of reactions is the conversion of six molecules of ribulose 5-phosphate into five molecules of glucose 6-phosphate.
6 Ribulose 5-phosphate+ H2O → 5 Glucose 6-phosphate + Pi
Finally, by summing all three groups of reactions, the oxidative phase, non-oxidative phase, and gluconeogenic steps, the overall reaction is:
Glucose 6-phosphate + 12 NADP+ + 7 H2O → 6 CO2 + 12 NADPH + 12 H+ + Pi
Thus, one molecule of glucose 6-phosphate, through six complete cycles of the pentose phosphate pathway (coupled with specific gluconeogenic reactions), is oxidized to six molecules of CO2. In the process, twelve molecules of NADPH are generated, with no net production of ribose 5-phosphate.[5][8][12]
When the need for NADPH and ATP is much greater than that for ribose 5-phosphate
When the cell requires significantly more NADPH than ribose 5-phosphate, and the energy charge is low, that is, there is a need for ATP, the ribulose 5-phosphate formed in the oxidative phase of the pentose phosphate pathway is converted into fructose 6-phosphate and glyceraldehyde 3-phosphate through the reactions of the non-oxidative phase. These two intermediates then enter glycolysis, where they are oxidized to pyruvate (the conjugate base of pyruvic acid), with the concomitant production of ATP.
The net reaction is as follows:
3 Glucose 6-phosphate + 6 NADP+ + 5 NAD+ + 5 Pi + 8 ADP → 5 Pyruvate + 3 CO2 + 6 NADPH + 5 NADH + 8 ATP + 2 H2O + 8 H+
If the cell requires additional ATP, the pyruvate produced can be further oxidized via the citric acid cycle. Conversely, if there is no need for further ATP production, the carbon skeleton of pyruvate can be diverted into various biosynthetic pathways.
Note: as in the previous scenario, there is no net production of ribose 5-phosphate.[5][8][12]
When the need for ribose 5-phosphate is much greater than that for NADPH
When significantly more ribose 5-phosphate than NADPH is required, as in rapidly dividing cells with high rates of nucleotide synthesis, precursors of DNA, the oxidative phase of the pentose phosphate pathway is bypassed, and NADPH is not produced. Instead, the rapid utilization of ribose 5-phosphate causes its intracellular levels to drop, thereby stimulating its synthesis. This is possible because the reactions of the non-oxidative phase are readily reversible.
In this metabolic context, most of the glucose 6-phosphate is first converted into fructose 6-phosphate and glyceraldehyde 3-phosphate via glycolysis. These intermediates are then used by transaldolase and transketolase to generate ribose 5-phosphate and xylulose 5-phosphate. The latter is further converted into ribose 5-phosphate through the actions of phosphopentose epimerase and ribose 5-phosphate isomerase.
The overall reaction is:
6 Glucose 6-phosphate + ATP → 6 Ribose 5-phosphate + ADP + H+
Under these metabolic conditions, there is a functional interplay between glycolysis and the non-oxidative phase of the pentose phosphate pathway, with the latter running in the direction of ribose 5-phosphate synthesis.
Note: no metabolites return to glycolysis in this case.[5][8][12]
When the needs for ribose 5-phosphate and NADPH are balanced
If the metabolic needs of the cell are satisfied by one molecule of ribose 5-phosphate and two molecules of NADPH per molecule of glucose 6-phosphate metabolized, the predominant reactions are those of the oxidative phase and the one catalyzed by ribose 5-phosphate isomerase.
The net reaction is:
Glucose 6-phosphate + 2 NADP+ + H2O → Ribose 5-phosphate + 2 NADPH + 2 H+ + CO2
Under these metabolic conditions as well, no metabolites return to glycolysis.[5][8][12]
| Scenario | Predominant pathway | Main output | Notes |
|---|---|---|---|
| High NADPH demand, low ribose 5-P or ATP need | Full oxidative phase + gluconeogenic recycling | ↑ NADPH (12 per G6P), CO2 (6), no net ribose 5-P | Supports fatty acid synthesis (e.g., in adipose tissue) |
| High NADPH and ATP demand, low ribose 5-P need | Oxidative phase + glycolysis | NADPH (6), ATP (8), pyruvate (5), NADH (5), CO2 (3) | Supports both biosynthesis and energy production |
| High ribose 5-P demand, low NADPH need | Non-oxidative phase from glycolytic intermediates | Ribose 5-P (6 per 6 G6P), consumes ATP | Found in rapidly dividing cells (e.g., cancer, bone marrow) |
| Balanced need for ribose 5-P and NADPH | Oxidative phase + ribose 5-P isomerase | Ribose 5-P (1), NADPH (2), CO2; no return to glycolysis | Typical of balanced anabolic demand |
Refences
- ^ a b c d e f Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
- ^ a b c d Rosenthal M.D., Glew R.H. Medical Biochemistry – Human Metabolism in Health and Disease. John Wiley J. & Sons, Inc., 2009.
- ^ Gebril H.M., Avula B., Wang Y.H., Khan I.A., Jekabsons M.B. (13)C metabolic flux analysis in neurons utilizing a model that accounts for hexose phosphate recycling within the pentose phosphate pathway. Neurochem Int 2016;93:26-39. doi:10.1016/j.neuint.2015.12.008
- ^ Otto Warburg – Nobel Lecture. NobelPrize.org. Nobel Prize Outreach 2025. Sun. 3 Aug 2025. https://www.nobelprize.org/prizes/medicine/1931/warburg/lecture/
- ^ a b c d e f g h i j k Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
- ^ Fritz Lipmann – Nobel Lecture. NobelPrize.org. Nobel Prize Outreach 2025. Sun. 3 Aug 2025. https://www.nobelprize.org/prizes/medicine/1953/lipmann/lecture/
- ^ a b Horecker B.L. The pentose phosphate pathway. J Biol Chem 2002;277(50):47965-47971. doi:10.1074/jbc.X200007200
- ^ a b c d e f g h i j k Garrett R.H., Grisham C.M. Biochemistry. 7th Edition. Cengage Learning, 2023.
- ^ a b c d Samland A.K., Sprenger G.A. Transaldolase: from biochemistry to human disease. Int J Biochem Cell Biol 2009;41(7):1482-1494. doi:10.1016/j.biocel.2009.02.001
- ^ a b c Michal G., Schomburg D. Biochemical pathways. An atlas of biochemistry and molecular biology. 2nd Edition. John Wiley & Sons, 2012.
- ^ Stincone A., Prigione A., Cramer T., Wamelink M.M., Campbell K., Cheung E., Olin-Sandoval V., Grüning N.M., Krüger A., Tauqeer Alam M., Keller M.A., Breitenbach M., Brindle K.M., Rabinowitz J.D., Ralser M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 2015;90(3):927-63. doi:10.1111/brv.12140
- ^ a b c d e f g h i j k Berg J.M., Tymoczko J.L., Gregory J.G. Jr., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
- ^ Sprenger G.A. Genetics of pentose-phosphate pathway enzymes of Escherichia coli K-12. Arch Microbiol 1995;164(5):324-30. doi:10.1007/BF02529978
- ^ a b c Au S.W.N., Gover S., Lam V.M.S., Adams M.J. Human glucose-6-phosphate dehydrogenase: the crystal structure reveals a structural NADP+ molecule and provides insights into enzyme deficiency. Structure 2000;8(3):293-303. doi:10.1016/S0969-2126(00)00104-0
- ^ a b c Harvey R.A., Ferrier D.R. Lippincott’s illustrated reviews: biochemistry. 5th Edition. Lippincott Williams & Wilkins, 2011.
- ^ Levy H.R. Glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides. Biochem Soc Trans. 1989 Apr;17(2):313-5. doi:10.1042/bst0170313
- ^ Cosgrove M.S., Naylor C., Paludan S., Adams M.J., Richard Levy H. On the mechanism of the reaction catalyzed by glucose 6-phosphate dehydrogenase. Biochemistry 1998;37(9);2759-2767. doi:10.1021/bi972069y
- ^ Stanton R.C. Glucose 6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012;64(5):362-9. doi:10.1002/iub.1017
- ^ a b c Patra K.C., Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci 2014;39(8):347-354. doi:10.1016/j.tibs.2014.06.005
- ^ García-Domínguez E., Carretero A., Viña-Almunia A., Domenech-Fernandez J., Olaso-Gonzalez G., Viña J., Gomez-Cabrera M.C. Glucose 6-P dehydrogenase – An antioxidant enzyme with regulatory functions in skeletal muscle during exercise. Cells 2022;11(19):3041. doi:10.3390/cells11193041
- ^ Phégnon L., Pérochon J., Uttenweiler-Joseph S., Cahoreau E., Millard P., Létisse F. 6-Phosphogluconolactonase is critical for the efficient functioning of the pentose phosphate pathway. FEBS J 2024;291(20):4459-4472. doi:10.1111/febs.17221
- ^ a b Ruda G.F., Campbell G., Alibu V.P., Barrett M.P., Brenk R., Gilbert I.H. Virtual fragment screening for novel inhibitors of 6-phosphogluconate dehydrogenase. Bioorg Med Chem 2010;18(14):5056-62. doi:10.1016/j.bmc.2010.05.077
- ^ a b Hanau S., Montin K., Cervellati C., Magnani M., Dallocchio F. 6-Phosphogluconate dehydrogenase mechanism: evidence for allosteric modulation by substrate. J Biol Chem 2010;285(28):21366-21371. doi:10.1074/jbc.M110.105601
- ^ Allard S.T., Giraud M.F., Naismith J.H. Epimerases: structure, function and mechanism. Cell Mol Life Sci 2001;58(11):1650-65. doi:10.1007/PL00000803
- ^ Samuel J., Tanner M.E. Mechanistic aspects of enzymatic carbohydrate epimerization. Nat Prod Rep 2002;19(3):261-77. doi:10.1039/b100492l
- ^ Bartkevihi L., Caruso Í.P., Martins B., Pires, J.R.M., Oliveira D.M.P., Anobom C.D., Almeida, F.C.L. Insights into the substrate uptake mechanism of Mycobacterium tuberculosis ribose 5-phosphate isomerase and perspectives on drug development. Biophysica 2023;3:139-157. doi:10.3390/biophysica3010010
- ^ Zhang R., Andersson C.E., Savchenko A., Skarina T., Evdokimova E., Beasley S., Arrowsmith C.H., Edwards A.M., Joachimiak A., Mowbray S.L. Structure of Escherichia coli ribose-5-phosphate isomerase: a ubiquitous enzyme of the pentose phosphate pathway and the Calvin cycle. Structure 2003;11(1):31-42. doi:10.1016/S0969-2126(02)00933-4
- ^ Kabashima T., Kawaguchi T., Wadzinski B.E., Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 2003;100:5107-5112. doi:10.1073/pnas.0730817100
- ^ Jelakovic S., Kopriva S., Süss K-H, Schulz G.E. Structure and catalytic mechanism of the cytosolic D-ribulose-5-phosphate 3-epimerase from rice. J Mol Biol 2003;326:127-135. doi:10.1016/S0022-2836(02)01374-8
- ^ a b c Kochetov G.A., Solovjeva O.N. Structure and functioning mechanism of transketolase. Biochim Biophys Acta 2014;1844(9):1608-1618. doi:10.1016/j.bbapap.2014.06.003
- ^ Solovjeva O.N. Regulation of enzymes with identical subunits on the example of transketolase. Open J Anal Bioanal Chem 2022;6(1):004-012. doi:10.17352/ojabc.000024
- ^ Sharkey T.D. Pentose phosphate pathway reactions in photosynthesizing cells. Cells 2021;10(6):1547. doi:10.3390/cells10061547
- ^ a b Lehwess-Litzmann A., Neumann P., Parthier C., Lüdtke S., Golbik R., Ficner R., Tittmann K. Twisted Schiff base intermediates and substrate locale revise transaldolase mechanism. Nat Chem Biol 2011;7(10):678-84. doi:10.1038/nchembio.633
- ^ Van Schaftingen E., Hers H-G. Inhibition of fructose-1,6-bisphosphatase by fructose-2,6-bisphosphate. Proc Natl Acad Sci USA 1981;78(5):2861-2863. doi:10.1073/pnas.78.5.2861
- ^ Van Schaftingen E., Jett M-F., Hue L., Hers H-G. Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc Natl Acad Sci USA 1981;78(6):3483-3486. doi:10.1073/pnas.78.6.3483