Nel 1956, Robert Sidney Cahn, Christopher Ingold, e Vladimir Prelog misero a punto un sistema di nomenclatura che, basandosi su poche e semplici regole, permette di assegnare la configurazione assoluta a ogni centro chirale presente in una molecola.[1][2]
Questo sistema di nomenclatura, chiamato sistema RS o convenzione di Cahn−Ingold−Prelog (CIP), quando accoppiato con il sistema di nomenclatura IUPAC, permette di assegnare in maniera accurata e priva di ambiguità il nome alle molecole chirali, anche quando è presente più di un centro asimmetrico.[3]
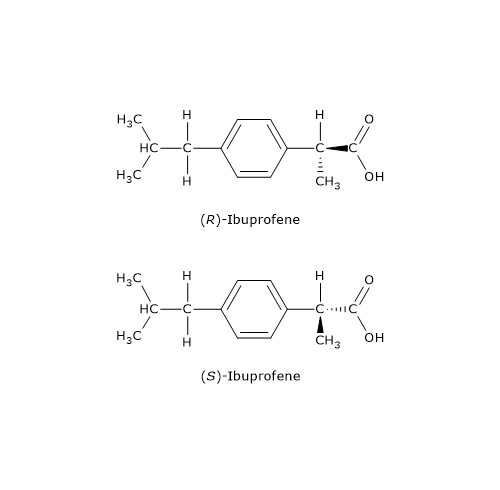
Le molecole chirali sono, nella maggior parte dei casi, in grado di far ruotare il piano della luce polarizzata quando questa attraversa una soluzione che le contenga. A questo riguardo va sottolineato che il segno del potere rotatorio non dà alcuna informazione circa la configurazione RS dei centri chirali della molecola.
La convenzione di Fischer−Rosanoff è un altro sistema di nomenclatura per le molecole chirali.[4] Tuttavia, rispetto al sistema RS, non analizza ogni singolo centro chirale ma assegna un nome all’intera molecola, e spesso presenta ambiguità con molecole con più centri chirali.[5]
In Sintesi: Punti Chiave
- Definizione: sistema di nomenclatura utilizzato per assegnare la configurazione assoluta ai centri chirali di una molecola.
- Regole di priorità: un insieme di tre regole utilizzate per determinare la configurazione R o S in base al numero atomico degli atomi legati, alla sequenza di atomi successivi e alla presenza di legami multipli.
- Contesto biochimico: consente la classificazione univoca di molecole con uno o più centri chirali, come amminoacidi e monosaccaridi.
- Indipendenza dall’attività ottica: la configurazione RS è un descrittore strutturale e non predice la direzione in cui una molecola ruota la luce polarizzata piana (+/−).
Indice
Regole di priorità del sistema RS
Il sistema RS assegna un ordine di priorità ai gruppi legati a un centro chirale e, tracciando una circonferenza dal gruppo a priorità maggiore verso quello a priorità minore, assegna la configurazione R o S al centro chirale.[6][7]
Prima regola
Si assegna un ordine di priorità ai gruppi, sulla base del numero atomico dell’atomo direttamente legato al centro chirale.
L’atomo con il numero atomico più alto ha la priorità più alta, mentre l’atomo con il numero atomico più basso ha la priorità più bassa.
Ad esempio, se un atomo di ossigeno (O, numero atomico 8), di carbonio (C, numero atomico 6), di cloro (Cl, numero atomico 17), e di bromo (Br, numero atomico 35) sono legati a un centro chirale, l’ordine di priorità sarà:
Br > Cl > O > C
Considerando gli isotopi, l’atomo con la massa atomica più alta ha la priorità più alta.[8]
Seconda regola
Quando gruppi differenti sono legati al centro chirale attraverso identici atomi, l’ordine di priorità è assegnato in base al numero atomico dell’atomo successivo a quello legato al centro, allontanandoci dal centro chirale finché non si raggiunge il primo punto di differenza.
Se, per esempio, i gruppi –CH3, –CH2CH3 e –CH2OH sono legati al centro di chiralità, ci sono tre atomi identici attaccati direttamente a esso. Analizzando gli atomi successivi, si ha:
- gruppo metilico (–CH3): H, H, H
- gruppo etilico (–CH2CH3): H, H, C
- gruppo idrossimetilico (–CH2OH): H, H, O
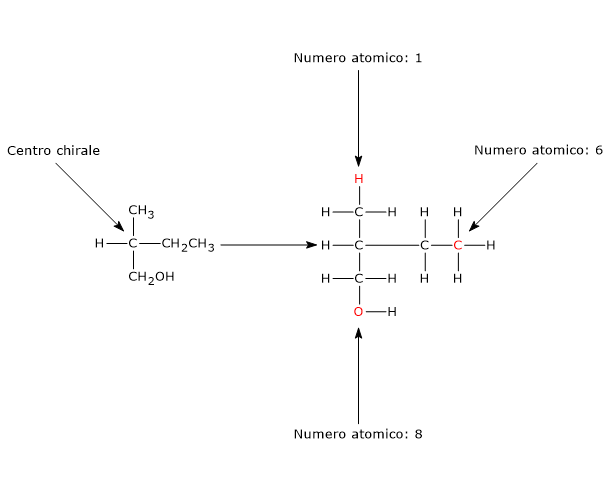
Poiché il numero atomico dell’atomo di ossigeno è maggiore di quello dell’atomo di carbonio, che a sua volta è maggiore di quello dell’atomo di idrogeno, l’ordine di priorità è: –CH2OH > –CH2CH3 > –CH3.
Per alcuni gruppi l’ordine di priorità è il seguente:
–I > –Br > –Cl > –SH > –OR > –OH > –NHR > –NH2 > –COOR > –COOH > –CHO > –CH2OH > –C6H5 > –CH3 > –2H > –1H
Nota: 2H e 1H si riferiscono rispettivamente agli isotopi deuterio e protio.
Si noti che i gruppi legati a un centro chirale devono avere un differente grado di priorità altrimenti il centro non può essere chirale.
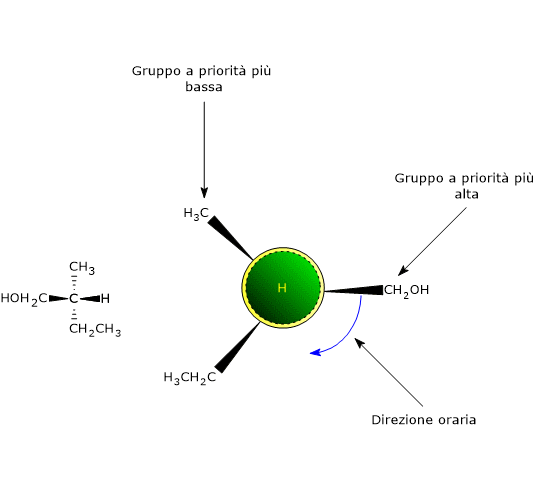
Determinazione della direzione oraria e antioraria
Stabilito l’ordine di priorità, si orienta la molecola nello spazio in modo che il gruppo a priorità più bassa sia diretto in direzione opposta a quella dell’osservatore, quindi dietro il centro chirale. A questo punto si congiungono i gruppi rimasti con una circonferenza, in modo che la successione sia secondo priorità decrescente: dal gruppo a priorità più alta verso quello a priorità più bassa.
- Se tracciando questa circonferenza si segue una direzione oraria, la configurazione del centro chirale è R, dal latino rectus che significa “destra”.
- Se tracciando la circonferenza si segue una direzione antioraria, la configurazione del centro chirale è S, dal latino sinister che significa “sinistra”.[8]
Terza regola
Questa può essere considerata come la terza regola del sistema RS, grazie alla quale è possibile assegnare la configurazione a un centro chirale anche quando sono presenti doppi o tripli legami nei gruppi legati al centro stesso.
Ai fini dell’attribuzione delle priorità, gli atomi impegnati nei legami multipli sono considerati come se fossero legati a un numero equivalente di atomi dello stesso tipo: sono duplicati nel caso di un doppio legame e triplicati nel caso di un triplo legame.
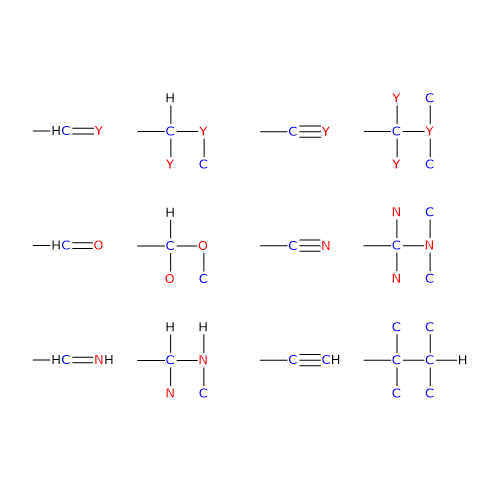
Nel caso di doppio legame C=Y, il carbonio è considerato legato a due atomi Y, e l’atomo Y è considerato legato a due carboni.
Nel caso di un triplo legame C≡Y, il carbonio è trattato come se fosse legato a tre atomi Y, e l’atomo Y come se fosse legato a tre carboni.[8]
Sistema RS in presenza di più centri chirali
Quando in una molecola sono presenti due o più centri chirali, si procede analizzando in modo indipendente ciascun centro, utilizzando le regole viste in precedenza.
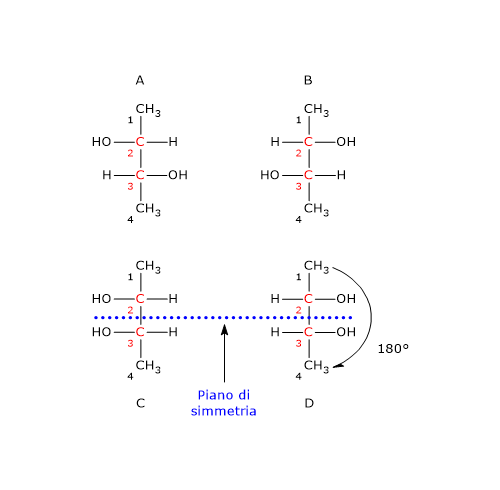
Consideriamo il 2,3-butandiolo. La molecola ha due centri chirali, il carbonio 2 e il carbonio 3, e tre stereoisomeri: due enantiomeri e un composto meso. Qual è la configurazione RS dei centri chirali dell’enantiomero in figura?
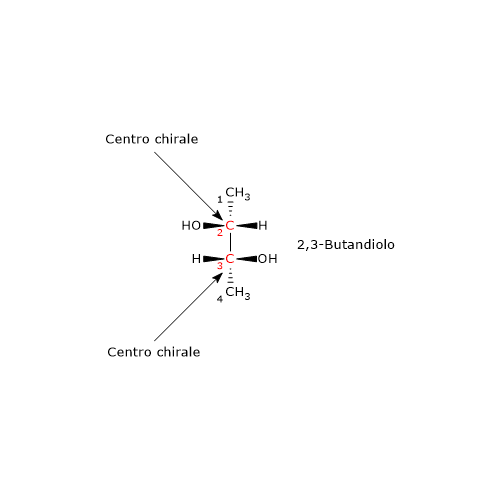
Si consideri il carbonio 2. L’ordine di priorità dei gruppi legati è:
–OH > –CH(OH)CH3 > –CH3 > –H
Si ruoti la molecola in modo che l’idrogeno, il gruppo con la priorità più bassa, sia diretto in direzione opposta all’osservatore. Disegnando una circonferenza partendo dal gruppo –OH, il gruppo con la priorità più alta, verso il gruppo –CH3, il gruppo a priorità più bassa, ci si muove in senso orario, per cui la configurazione del carbonio 2 è R.
Applicando la stessa procedura al carbonio 3, si scopre che la sua configurazione è R.
Quindi l’enantiomero in figura è il (2R,3R)-2,3-butandiolo.
Amminoacidi e gliceraldeide
Nella convenzione di Fischer-Rosanoff tutti gli amminoacidi proteinogenici sono L-amminoacidi. Nel sistema RS, con l’eccezione della glicina che non è chirale, e della cisteina che, per la presenza di un gruppo tiolico, è classificata come (R)-cisteina, tutti gli altri amminoacidi proteinogenici sono (S)-amminoacidi.
La treonina e l’isoleucina posseggono due centri chirali, il carbonio α e un atomo di carbonio sulla catena laterale, e tre stereoisomeri: due enantiomeri e un composto meso. Le forme dei due amminoacidi isolate dalle proteine sono la:
-
- (2S,3R)-treonina;
- (2S,3S)-isoleucina.
Secondo la convenzione di Fischer-Rosanoff queste corrispondono rispettivamente alla L-treonina e alla L-isoleucina.
Nel sistema RS, la L-gliceraldeide ha il centro chirale con configurazione S, per cui è indicata come (S)-gliceraldeide; ovviamente la D-gliceraldeide sarà la (R)-gliceraldeide.[9]
Bibliografia
- ^ Cahn R.S., Ingold C., Prelog V. Specification of molecular chirality. Angew Chem 1966:5(4); 385-415. doi:10.1002/anie.196603851
- ^ Prelog V. and Helmchen G. Basic principles of the CIP‐system and proposals for a revision. Angew Chem 1982:21(8);567-583. doi:10.1002/anie.198205671
- ^ IUPAC. Compendium of chemical terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. doi:10.1351/goldbook
- ^ Rosanoff M.A. On Fischer’s classification of stereo-isomers. J Am Chem Soc 1906:28(1);114-121. doi:10.1021/ja01967a014
- ^ Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
- ^ Garrett R.H., Grisham C.M. Biochemistry. 7th Edition. Cengage Learning, 2023.
- ^ Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of biochemistry. 5th Edition. Pearson, 2012.
- ^ a b c Solomons T.W.G., Fryhle C.B., Snyder S.A. Solomons’ organic chemistry. 12th Edition. John Wiley & Sons Incorporated, 2017.
- ^ Morris D.G., Drayton C., Hepworth J.D. Stereochemistry. Royal society of chemistry. 2001. doi:10.1039/9781847551948