Gli enzimi multifunzionali sono proteine in cui due o più siti attivi che catalizzano reazioni consecutive di una via metabolica sono presenti in una catena polipeptidica. Sembra probabile che derivino da eventi di fusione genica rappresentando, al pari dei complessi multienzimatici, una soluzione adottata dall’evoluzione per massimizzare l’efficienza catalitica, assicurando vantaggi che non si avrebbero nel caso in cui le singole attività enzimatiche fossero presenti su proteine distinte e libere.[1][2]
Indice
Vantaggi degli enzimi multifunzionali
Gli organismi viventi combattono con l’inevitabile processo di decadimento che, se non contrastato, conduce a un crescente aumento del disordine, sino alla morte.
A livello molecolare il mantenimento della vita è reso possibile dalla grande efficienza raggiunta dagli enzimi nell’accelerare le reazioni chimiche e nell’evitare reazioni collaterali. Un’idea della velocità con cui procede il metabolismo cellulare è fornita dalla velocità del turn over dell’ATP in una cellula di mammifero: ogni 1–2 minuti l’intero pool del trifosfato è idrolizzato e risintetizzato. Tradotto in numeri questo corrisponde al turn over di circa 107 molecole di ATP al secondo, e, per il corpo umano, a circa 1 grammo di ATP al minuto.[3] Alcuni enzimi hanno addirittura raggiunto la perfezione catalitica, ossia sono talmente efficienti che quasi ogni collisione con il proprio substrato porta alla catalisi.[2]
Uno dei fattori limitanti la velocità di una reazione enzimatica è la proprio frequenza di collisione tra enzimi e substrati. Il modo più semplice per aumentare tale frequenza sarebbe quello di aumentare la concentrazione di substrati ed enzimi. Tuttavia, dato l’elevatissimo numero di differenti reazioni che avvengono all’interno della cellula, questa strada non è praticabile. Esiste cioè un limite fisico alle concentrazioni che substrati ed enzimi possono raggiungere, concentrazioni che sono dell’ordine delle micromoli per i substrati, e anche più basse per gli enzimi.[3] Fanno eccezione gli enzimi della glicolisi nelle cellule muscolari e negli eritrociti, dove raggiungono concentrazioni dell’ordine delle 0.1 mM e anche maggiori.[4]
Incanalamento dei substrati
Una delle strade percorse dall’evoluzione per aumentare la velocità con cui procedono le reazioni enzimatiche è stata quella di selezionare strutture molecolari, quali gli enzimi multifunzionali e i complessi multienzimatici, che permettano, attraverso l’ottimizzazione dell’organizzazione spaziale degli enzimi di una via metabolica, di minimizzare la distanza che il prodotto della reazione A deve percorrere per raggiungere il sito attivo che catalizza la reazione successiva B, e così via, ottenendo cioè l’incanalamento dei substrati, in inglese substrate channeling, della via stessa.[5] Per alcuni enzimi multifunzionali e complessi multienzimatici l’incanalamento è ottenuto grazie alla presenza di veri e propri tunnel intramolecolari.[6]
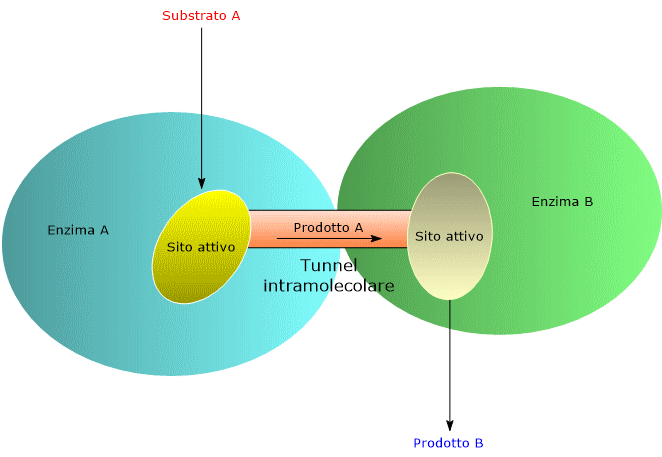 L’incanalamento dei substrati migliora l’efficienza catalitica, e quindi la velocità di reazione, in vari modi, di seguito brevemente descritti.
L’incanalamento dei substrati migliora l’efficienza catalitica, e quindi la velocità di reazione, in vari modi, di seguito brevemente descritti.
- Minimizza la diffusione nel mezzo circostante del reagente, quindi la sua diluizione. Questo consente di raggiungere concentrazioni locali elevate, anche quando la sua concentrazione nella cellula è bassa, aumentando quindi la frequenza delle collisioni enzima-substrato.
- Riduce il tempo di transito dei substrati da un sito attivo al successivo.
- Minimizza la probabilità che si verifichino reazioni collaterali.
- Riduce la probabilità che intermedi chimicamente labili siano degradati.[2][5]
Gli enzimi multifunzionali offrono vantaggi anche dal punto di vista della regolazione della loro sintesi, essendo possibile coordinare la sintesi di tutte le attività enzimatiche interessate grazie al fatto che la struttura è codificata da un singolo gene.[7]
Infine, analogamente ai complessi multienzimatici, le attività enzimatiche presenti possono essere regolate in modo coordinato. Inoltre, poiché spesso l’enzima che catalizza la tappa di comando della sequenza coincide con quello che catalizza la prima reazione, si evita la sintesi di molecole non necessarie, che verrebbero prodotte se la tappa di comando fosse posta a valle, prevenendo così sia lo spreco di energia sia la sottrazione di metaboliti ad altre vie metaboliche.[2]
Esempi di enzimi multifunzionali
Al pari dei complessi multienzimatici, anche gli enzimi multifunzionali sono molto comuni e coinvolti in vie metaboliche sia anaboliche che cataboliche.
Di seguito alcuni esempi.
Acetil-CoA carbossilasi
L’acetil-CoA carbossilasi (ACC; EC 6.4.1.2), una carbossilasi biotina-dipendente, è formata da due enzimi, la biotina carbossilasi (EC 6.3.4.14) e una transcarbossilasi, più la proteina trasportatrice della biotina (BCCP). ACC catalizza la sintesi del malonil-CoA via carbossilazione dell’acetil-CoA, reazione che è la tappa di comando della sintesi degli acidi grassi.[7] La sintesi del maloni-CoA procede in due tappe.
- Nella prima la biotina carbossilasi catalizza il trasferimento di una molecola di anidride carbonica (CO2) dallo ione bicarbonato a un atomo di azoto dell’anello della biotina, che funge da trasportatore temporaneo di CO2. La reazione comporta il consumo di una molecola di ATP.
- Nella seconda tappa la transcarbossilasi catalizza il trasferimento del gruppo carbossilico dalla carbossi-biotina all’acetil-CoA a dare malonil-CoA.
Il malonil-CoA verrà di seguito utilizzato come donatore di unità bicarboniose dalla acido grasso sintasi (EC 2.3.1.85) durante l’allungamento degli acidi grassi.[8]
Nei mammiferi e negli uccelli l’acetil-CoA carbossilasi è un enzima multifunzionale essendo le due attività enzimatiche, e BCCP, presenti su un’unica catena polipeptidica.[9]
Nei batteri invece ACC è un complesso multienzimatico formato dall’aggregazione di tre catene polipeptidiche, ossia i due enzimi più BCCP.[10]
Nelle piante superiori sono presenti entrambe le forme.[8]
Acido grasso sintasi di tipo I
L’acido grasso sintasi (FAS) catalizza la sintesi dell’acido palmitico utilizzando il malonil-CoA, che è il prodotto della reazione catalizzata dalla acetil-CoA carbossilasi, come donatore di unità bicarboniose.
Esistono due tipi di FAS.
- Negli animali e nei funghi è un enzima multifunzionale, ed è detto di Tipo I.
Negli animali è un omodimero; in ogni catena polipeptidica sono presenti le sette le attività enzimatiche e la proteina trasportatrice di acili (ACP).
Nei lieviti e nei funghi FAS è formata da due subunità multifunzionali, dette α and β, disposte a formare una struttura eterododecamerica α6β6. - Nella maggior parte dei procarioti e nelle piante l’acido grasso sintasi, detta di Tipo II, non è un enzima multifunzionale ma un complesso multienzimatico essendo composto da enzimi distinti più ACP.[11][12][13]
PRA isomerasi:IGP sintasi
La biosintesi dell’aminoacido triptofano a partire dal corismato coinvolge diversi passaggi, di seguito brevemente descritti.
- Nel primo, la glutammina dona un atomo di azoto all’anello indolico del corismato, che è convertito in antranilato, e la glutammina in glutammato; la reazione è catalizzata dalla antranilato sintasi (EC 4.1.3.27).
- L’antranilato è fosforibosilato a spese del 5-fosforibosil-1-pirofosfato o PRPP a dare N-(5’-fosforibosil)-antranilato (PRA), nella reazione catalizzata dalla antranilato fosforibosiltransferasi (EC 2.4.2.18).
- Nel passaggio successivo, catalizzato dalla PRA isomerasi (EC 5.3.1.24), PRA è isomerizzato a dare enol-1-o-carbossifenilamino-1-desossiribulosio fosfato (CdRP). PRA e CdRP sono un esempio di isomeria di struttura.
- CdRP è convertito in indolo-3-glicerolo fosfato (IGP) nella reazione catalizzata dalla indolo-3-glicerofosfato sintasi (IGP) sintasi (EC 4.1.1.48).
- Infine, la triptofano sintasi (EC 4.2.1.20) catalizza gli ultimi due passaggi della via: la conversione di IGP in indolo, una idrolisi, e la reazione dell’indolo con una serina a dare il triptofano.[11]
In E. coli, PRA isomerasi e IGP sintasi sono presenti su un’unica catena polipeptidica, che è quindi un enzima bifunzionale.[14] In altri microorganismi, come Bacillus subtilis, Salmonella typhimurium e Pseudomonas putida le due attività enzimatiche sono presenti su catene polipeptidiche distinte.[2] La triptofano sintasi è invece un esempio di complesso multienzimatico, e uno degli esempi meglio caratterizzati di canalizzazione del flusso dei metaboliti.[15][16]
Glutammina-PRPP amido transferasi
La glutammina-PRPP ammidotransferasi (EC 2.4.2.14) catalizza la prima della 10 tappe che portano alla sintesi de novo delle purine, ossia la formazione della 5-fosforibosilammina a seguito del trasferimento dell’azoto ammidico della glutammina, che funge quindi da fonte di azoto, al PRPP.
La reazione procede in due tappe, catalizzate nei due siti attivi dell’enzima, un sito N-terminale e uno C-terminale.
- Nella prima tappa, il sito attivo N-terminale catalizza l’idrolisi dell’azoto ammidico della glutammina a dare ammoniaca e glutammato.
- Nella seconda tappa, catalizzata dal sito attivo C-terminale, che ha attività fosforibosiltransferasica, l’ammoniaca precedentemente rilasciata viene legata al C-1 del PRPP a dare la 5-fosforibosilammina. In questa reazione si verifica l’inversione della configurazione del C-1 del ribosio, da α a β, stabilendo la forma anomerica del nucleotide in via di sintesi.
Ci sono tre punti di controllo che cooperano nella regolazione della sintesi de novo dei nucleotidi purinici, e la reazione catalizzata dalla glutammina-PRPP ammidotransferasi, che è anche la prima reazione esclusiva della via, è il primo.[2]
Al pari del complesso della carbamil fosfato sintetasi batterica (EC 6.3.4.16), anche i siti attivi di questo enzima multifunzionale sono connessi attraverso un canale intramolecolare. Tuttavia questo canale è più corto, essendo lungo circa 20 Å, ed è delimitato da residui amminoacidici non polari, quindi è altamente idrofobico. Mancando gruppi in grado di stabilire legami idrogeno, il tunnel permette la diffusione dell’ammoniaca verso il secondo sito attivo.[17][18]
CAD
La sintesi de novo dei nucleotidi pirimidinici avviene attraverso una serie di reazioni enzimatiche che, a differenza di quanto accade nella sintesi de novo dei nucleotidi purinici, comporta dapprima la formazione dell’anello pirimidinico e quindi il suo legame al ribosio-5-fosfato. Le prime tre tappe della via sono catalizzate in sequenza dalla carbamil fosfato sintetasi, aspartato transcarbamilasi (EC 2.1.3.2) e diidroorotasi (EC 3.5.2.3), e sono comuni a tutte le specie.
- Nella prima tappa, la carbamil fosfato sintetasi, che presenta due attività enzimatiche, ossia una attività amidotransferasica glutammina dipendente e un’attività sintasica, catalizza la sintesi del carbamil fosfato a partire da glutammina, ione bicarbonato e ATP.
- Nella seconda tappa, che è quella di comando della via metabolica ed è catalizzata dalla aspartato transcarbamilasi, il carbamil fosfato reagisce con l’aspartato a dare l’N-carbamil aspartato.
- Infine la diidroorotasi, catalizzando la rimozione di una molecola d’acqua dall’N-carbamil aspartato, porta alla chiusura dell’anello pirimidinico a formare L-diidroorotato.[8]
Negli eucarioti, in particolare nei mammiferi, in Drosofila e Dictyostelium, un genere di amebe, le tre attività sono presenti su una singola catena polipeptidica, codificata da un singolo gene derivante da una fusione avvenuta almeno 100 milioni di anni fa. L’enzima multifunzionale, abbreviato come CAD, è un omomultimero composto da tre subunità o più.[19]
Nei procarioti invece i tre enzimi sono indipendenti, e la carbamil fosfato sintetasi è un esempio di complesso multienzimatico.[20]
Nei lieviti l’attività diidroorotasica è presente su una proteina separata.[21]
È interessante notare che gli studi sull’attività enzimatica hanno rivelato evidenze di un incanalamento dei substrati, in particolare tra i primi due passaggi. Questo incanalamento sembra essere più efficiente nella proteina del lievito rispetto all’enzima CAD dei mammiferi.[22]
Bibliografia
- ^ Pareek V., Sha Z., He J., Wingreen N.S., Benkovic S.J. Metabolic channeling: predictions, deductions, and evidence. Mol Cell 2021;81(18):3775-3785. doi:10.1016/j.molcel.2021.08.030
- ^ a b c d e f Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
- ^ a b Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. 6th Edition. Garland Science, Taylor & Francis Group, 2015.
- ^ Maughan D.W., Henkin J.A., Vigoreaux J.O. Concentrations of glycolytic enzymes and other cytosolic proteins in the diffusible fraction of a vertebrate muscle proteome. Mol Cell Proteomics 2005;4(10):1541-9. doi:10.1074/mcp.M500053-MCP200
- ^ a b Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
- ^ Kondrat S., von Lieres E. Mechanisms and effects of substrate channelling in enzymatic cascades. Methods Mol Biol 2022;2487:27-50. doi:10.1007/978-1-0716-2269-8_3
- ^ a b Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002.
- ^ a b c Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
- ^ Wang Y., Yu W., Li S., Guo D., He J., Wang Y. Acetyl-CoA carboxylases and diseases. Front Oncol 2022;12:836058. doi:10.3389/fonc.2022.836058
- ^ Salie M.J., Thelen J.J. Regulation and structure of the heteromeric acetyl-CoA carboxylase. Biochim Biophys Acta 2016;1861(9 Pt B):1207-1213. doi:10.1016/j.bbalip.2016.04.004
- ^ a b Garrett R.H., Grisham C.M. Biochemistry. 4th Edition. Brooks/Cole, Cengage Learning, 2010.
- ^ Günenc A.N., Graf B., Stark H., Chari A. Fatty acid synthase: structure, function, and regulation. Subcell Biochem 2022;99:1-33. doi:10.1007/978-3-031-00793-4_1
- ^ Wakil S.J. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry 1989;28(11):4523-30. doi:10.1021/bi00437a001
- ^ Priestle J.P., Grütter M.G., White J.L., Vincent M.G., Kania M., Wilson E., Jardetzky T.S., Kirschner K., Jansonius J.N. Three-dimensional structure of the bifunctional enzyme N-(5′-phosphoribosyl)anthranilate isomerase-indole-3-glycerol-phosphate synthase from Escherichia coli. Proc Natl Acad Sci USA 1987;84(16):5690-4. doi:10.1073/pnas.84.16.5690
- ^ Hyde C.C., Ahmed S.A., Padlan E.A., Miles E.W., and Davies D.R. Three-dimensional structure of the tryptophan synthase multienzyme complex from Salmonella typhimurium. J Biol Chem 1988:263(33);17857-17871. doi:10.1016/S0021-9258(19)77913-7
- ^ Hyde C.C., Miles E.W. The tryptophan synthase multienzyme complex: exploring structure-function relationships with X-ray crystallography and mutagenesis. Nat Biotechnol 1990:8;27-32. doi:10.1038/nbt0190-27
- ^ Muchmore C.R.A., Krahn J.M, Smith J.L., Kim J.H., Zalkin H. Crystal structure of glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Protein Sci 1998:7;39-51. doi:10.1002/pro.5560070104
- ^ Raushel F.M., Thoden J.B., Holden H.M. The amidotransferase family of enzymes: molecular machines for the production and delivery of ammonia. Biochemistry 1999;38(25):7891-9. doi:10.1021/bi990871p
- ^ Del Caño-Ochoa F., Moreno-Morcillo M., Ramón-Maiques S. CAD, a multienzymatic protein at the head of de novo pyrimidine biosynthesis. Subcell biochem 2019;93:505-538. doi:10.1007/978-3-030-28151-9_17
- ^ Washabaugh M.W., Collins K.D. Dihydroorotase from Escherichia coli. Purification and characterization. J Biol Chem 1984;259(5):3293-8.
- ^ Guan H.H., Huang Y.H., Lin E.S., Chen C.J., Huang C.Y. Structural analysis of Saccharomyces cerevisiae dihydroorotase reveals molecular insights into the tetramerization mechanism. Molecules 2021;26(23):7249. doi:10.3390/molecules26237249
- ^ Belkaïd M., Penverne B., Hervé G. In situ behavior of the pyrimidine pathway enzymes in Saccharomyces cerevisiae. 3. Catalytic and regulatory properties of carbamylphosphate synthetase: channeling of carbamylphosphate to aspartate transcarbamylase. Arch Biochem Biophys 1988;262(1):171-80. doi:10.1016/0003-9861(88)90179-8