Multifunctional enzymes are proteins in which two or more enzymatic activities, catalyzing consecutive steps of a metabolic pathway, are located on the same polypeptide chain. They are thought to have arisen through gene fusion events and, like multienzyme complexes, represent an evolutionary strategy to maximize catalytic efficiency. This configuration provides advantages that would not be possible if these enzymatic activities were present on separate proteins freely dissolved in the cytosol.[1][2]
Contents
What advantages do multifunctional enzymes provide?
Living organisms constantly fight against natural processes of decay which, if left unchecked, lead to increasing disorder and ultimately death.
At the molecular level, life is sustained by the extraordinary effectiveness of enzymes in accelerating chemical reactions and minimizing side reactions. The rate of ATP turnover in a mammalian cell gives an idea of how fast cellular metabolism operates: every 1–2 minutes, the entire ATP pool is turned over, hydrolyzed and then regenerated via phosphorylation. This corresponds to a turnover of about 107 ATP molecules per second, and, for the human body, to about 1 gram of ATP every minute.[3] Some enzymes have even achieved catalytic perfection, meaning they are so efficient that nearly every collision with their substrate leads to catalysis.[2]
One of the limiting factors of an enzymatic reaction is the frequency of collisions between enzymes and substrates. The simplest way to increase this frequency would be to increase their concentrations. However, given the vast number of different reactions taking place within the cell, this strategy is not feasible. In other words, there is a physical limit to the concentrations that substrates and enzymes can reach, substrate concentrations which are typically in the micromolar range, and even lower for enzymes.[3] Exceptions include the enzymes of glycolysis in muscle cells and erythrocytes, which can be present at concentrations around 0.1 mM or even higher.[4]
Metabolic channeling
One of the strategies favored by evolution to increase the rate of enzymatic reactions is the selection of molecular structures, such as multifunctional enzymes and multienzyme complexes, that optimize the spatial organization of enzymes within a metabolic pathway. This spatial optimization minimizes the distance that the product of reaction A must travel to reach the active site catalyzing reaction B in the sequence, and so on. This phenomenon is known as substrate channeling, or more broadly, metabolic channeling.[5] In some multifunctional enzymes and multienzyme complexes, channeling is achieved through the presence of intramolecular channels.[6]
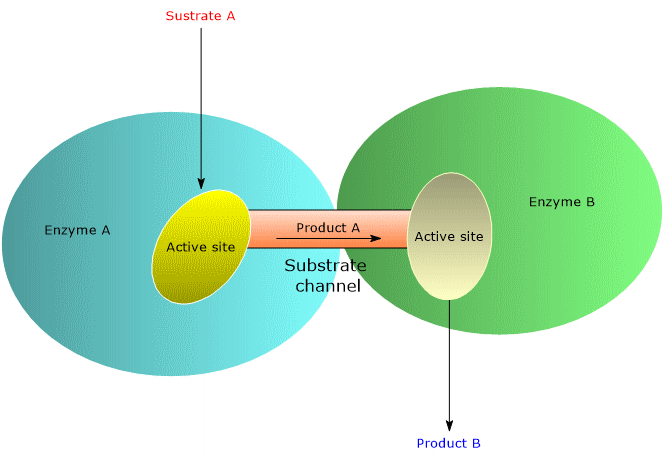 Metabolic channeling enhances catalytic efficiency, and therefore the reaction rate, in several ways, outlined below.
Metabolic channeling enhances catalytic efficiency, and therefore the reaction rate, in several ways, outlined below.
- It reduces the diffusion of substrates into the bulk solvent, thereby minimizing their dilution. This enables high local substrate concentrations, even when their overall cellular concentrations are low, increasing the frequency of enzyme-substrate collisions.
- It reduces the time required for substrates to diffuse from one active site to the next.
- It lowers the probability of side reactions.
- It decreases the likelihood that unstable intermediates will degrade before reaching the next active site.[2][5]
Multifunctional enzymes also offer advantages in terms of the regulation of their synthesis. Since all enzymatic activities are encoded by a single gene, their expression can be coordinated efficiently.[7]
Finally, like multienzyme complexes, multifunctional enzymes enable coordinated control of catalytic activities. Often, the enzyme that catalyzes the committed step of the pathway is also responsible for the first reaction. This positioning prevents the synthesis of unnecessary products, which would occur if regulation took place downstream, thereby avoiding waste of energy and preventing the depletion of metabolites from other metabolic pathways.[2]
Examples of multifunctional enzymes
Like multienzyme complexes, multifunctional enzymes are also common and play key roles in many metabolic pathways, both anabolic and catabolic.
Below are several noteworthy examples.
Acetyl-CoA carboxylase
Acetyl-CoA carboxylase (ACC; EC 6.4.1.2) is a biotin-dependent carboxylase composed of two catalytic domains, biotin carboxylase (EC 6.3.4.14) and carboxyltransferase, as well as a biotin carboxyl carrier protein (BCCP). ACC catalyzes the synthesis of malonyl-CoA by carboxylating acetyl-CoA, in a reaction that represents the committed step in fatty acid biosynthesis.[7] The reaction occurs in two sequential steps.
- In the first step, biotin carboxylase catalyzes the ATP-dependent carboxylation of the nitrogen atom in biotin, which acts as a carrier of carbon dioxide (CO2). The source of CO2 in this reaction is the bicarbonate ion.
- In the second step, carboxyltransferase catalyzes the transfer of the carboxyl group from carboxybiotin to acetyl-CoA, forming malonyl-CoA.
Malonyl-CoA then serves as the donor of an activated two-carbon unit to fatty acid synthase (EC 2.3.1.85) during the elongation of fatty acid chains.[8]
In mammals and birds, acetyl-CoA carboxylase is a multifunctional enzyme, with biotin carboxylase, carboxyltransferase, and BCCP activities located on a single polypeptide chain.[9]
In contrast, in bacteria, ACC exists as a multienzyme complex composed of three distinct polypeptide chains: biotin carboxylase, carboxyltransferase, and BCCP.[10]
Both forms, the multifunctional enzyme and the multienzyme complex, are found in higher plants.[8]
Type I fatty acid synthase
Fatty acid synthase (FAS) catalyzes the synthesis of palmitic acid, using malonyl-CoA, the product of the reaction catalyzed by acetyl-CoA carboxylase, as a donor of two-carbon units.
There are two types of FAS.
- In fungi and animals, FAS is a multifunctional enzyme known as Type I.
In animals, it exists as a homodimer, with each polypeptide chain containing all seven enzymatic activities, along with an acyl carrier protein (ACP) domain.
In yeast and fungi, FAS is composed of two distinct multifunctional subunits, designated α and β, which assemble into an α6β6 heterododecameric complex. - In most prokaryotes and in plants, FAS is classified as Type II. It is not a multifunctional enzyme, but rather a multienzyme complex composed of separate, individual enzymes and ACP.[11][12][13]
PRA-isomerase:IGP synthase
The biosynthesis of the amino acid tryptophan from chorismate involves several steps, summarized below.
- In the first step, glutamine donates a nitrogen atom to the indole ring of chorismate, which is converted into anthranilate, while glutamine is simultaneously converted into glutamate. This reaction is catalyzed by anthranilate synthase (EC 4.1.3.27).
- Anthranilate is then phosphoribosylated to form N-(5′-phosphoribosyl)-anthranilate (PRA), in a reaction catalyzed by anthranilate phosphoribosyltransferase (EC 2.4.2.18).
In this step, 5-phosphoribosyl-1-pyrophosphate (PRPP) acts as the donor of a 5-phosphoribosyl group. - In the next step, PRA is isomerized to enol-1-o-carboxyphenylamino-1-deoxyribulose phosphate (CdRP) by PRA isomerase (EC 5.3.1.24).
PRA and CdRP are an example of structural isomer. - CdRP is then converted into indole-3-glycerol phosphate (IGP) in a reaction catalyzed by indole-3-glycerol phosphate synthase (IGP synthase) (EC 4.1.1.48).
- Finally, tryptophan synthase (EC 4.2.1.20) catalyzes the last two steps of the pathway: the hydrolysis of IGP to indole, and the condensation of indole with serine to form tryptophan.[11]
In E. coli, PRA isomerase and IGP synthase are located on a single polypeptide chain, making it a bifunctional enzyme.[14] In other microorganisms, such as Bacillus subtilis, Salmonella typhimurium, and Pseudomonas putida, the two catalytic activities are found on separate polypeptide chains.[2] In contrast, tryptophan synthase is a classic example of a multienzyme complex, and one of the best-characterized examples of metabolic channeling.[15][16]
Glutamine-PRPP amidotransferase
Glutamine-PRPP amidotransferase (GPATase; EC 2.4.2.14) catalyzes the first of ten steps in the de novo synthesis of purine nucleotides, specifically, the formation of 5-phosphoribosylamine via transfer of the amide nitrogen from glutamine to PRPP.
In this reaction, glutamine acts as a nitrogen donor.
The reaction proceeds in two steps, which occur at distinct active sites: one located at the N-terminal and the other at the C-terminal region of the enzyme.
- In the first step, the N-terminal active site catalyzes the hydrolysis of glutamine, releasing glutamate and ammonia.
- In the second step, catalyzed by the C-terminal active site (which has phosphoribosyltransferase activity), the ammonia is attached to the C-1 position of PRPP, forming 5-phosphoribosylamine.
This step also involves inversion of configuration at the ribose C-1 carbon, from α to β, thereby establishing the anomeric form of the future nucleotide.
There are three key regulatory points in the control of purine nucleotide biosynthesis. The reaction catalyzed by GPATase, being the first committed step of the pathway, represents the primary control point.[2]
As in the bacterial carbamoyl phosphate synthetase complex (EC 6.3.4.16), the active sites of this multifunctional enzyme are connected by an intramolecular channel. However, in this case the channel is shorter, approximately 20 Å in length, and is lined with conserved nonpolar (hydrophobic) amino acids.
Due to the lack of hydrogen-bonding groups, the channel does not impede the diffusion of ammonsia between active site.[17][18]
CAD
The de novo synthesis of pyrimidine nucleotides proceeds through a series of enzymatic reactions that, unlike the de novo synthesis of purine nucleotides, begins with the formation of the pyrimidine ring, which is then attached to ribose 5-phosphate. The first three steps of the pathway are catalyzed sequentially by the enzymes carbamoyl phosphate synthetase, aspartate transcarbamoylase (EC 2.1.3.2), and dihydroorotase (EC 3.5.2.3), a sequence that is conserved across all species.
- In the first step, carbamoyl phosphate synthetase, which has two enzymatic activities, a glutamine-dependent amidotransferase and a synthase, catalyzes the formation of carbamoyl phosphate from glutamine, bicarbonate ion, and ATP.
- In the second step, catalyzed by aspartate transcarbamoylase, carbamoyl phosphate reacts with aspartate to form N-carbamoyl aspartate. This is the committed step of the pathway.
- In the final step, dihydroorotase catalyzes the removal of water from N-carbamoyl aspartate, leading to ring closure and the formation of L-dihydroorotate.[8]
In eukaryotes, particularly in mammals, Drosophila, and Dictyostelium (a genus of amoebae), these three enzymatic activities are located on a single polypeptide chain, encoded by a gene derived from a gene fusion event that occurred at least 100 million years ago. The resulting multifunctional enzyme, known by the acronym CAD, functions as a homomultimer composed of three or more subunits.[19]
In contrast, in prokaryotes, the three enzymes exist as separate proteins, and carbamoyl phosphate synthetase is a typical example of a multienzyme complex.[20]
In yeasts, dihydroorotase is located on a distinct polypeptide chain, separate from the other two enzymes.[21]
Interestingly, studies of enzymatic activity have revealed evidence of substrate channeling, particularly between the first two steps. This channeling appears to be more efficient in the yeast protein compared to the mammalian CAD enzyme.[22]
References
- ^ Pareek V., Sha Z., He J., Wingreen N.S., Benkovic S.J. Metabolic channeling: predictions, deductions, and evidence. Mol Cell 2021;81(18):3775-3785. doi:10.1016/j.molcel.2021.08.030
- ^ a b c d e f Voet D. and Voet J.D. Biochemistry. 4th Edition. John Wiley J. & Sons, Inc. 2011.
- ^ a b Alberts B., Johnson A., Lewis J., Morgan D., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. 6th Edition. Garland Science, Taylor & Francis Group, 2015.
- ^ Maughan D.W., Henkin J.A., Vigoreaux J.O. Concentrations of glycolytic enzymes and other cytosolic proteins in the diffusible fraction of a vertebrate muscle proteome. Mol Cell Proteomics 2005;4(10):1541-9. doi:10.1074/mcp.M500053-MCP200
- ^ a b Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
- ^ Kondrat S., von Lieres E. Mechanisms and effects of substrate channelling in enzymatic cascades. Methods Mol Biol 2022;2487:27-50. doi:10.1007/978-1-0716-2269-8_3
- ^ a b Berg J.M., Tymoczko J.L., and Stryer L. Biochemistry. 5th Edition. W. H. Freeman and Company, 2002.
- ^ a b c Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
- ^ Wang Y., Yu W., Li S., Guo D., He J., Wang Y. Acetyl-CoA carboxylases and diseases. Front Oncol 2022;12:836058. doi:10.3389/fonc.2022.836058
- ^ Salie M.J., Thelen J.J. Regulation and structure of the heteromeric acetyl-CoA carboxylase. Biochim Biophys Acta 2016;1861(9 Pt B):1207-1213. doi:10.1016/j.bbalip.2016.04.004
- ^ a b Garrett R.H., Grisham C.M. Biochemistry. 4th Edition. Brooks/Cole, Cengage Learning, 2010.
- ^ Günenc A.N., Graf B., Stark H., Chari A. Fatty acid synthase: structure, function, and regulation. Subcell Biochem 2022;99:1-33. doi:10.1007/978-3-031-00793-4_1
- ^ Wakil S.J. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry 1989;28(11):4523-30. doi:10.1021/bi00437a001
- ^ Priestle J.P., Grütter M.G., White J.L., Vincent M.G., Kania M., Wilson E., Jardetzky T.S., Kirschner K., Jansonius J.N. Three-dimensional structure of the bifunctional enzyme N-(5′-phosphoribosyl)anthranilate isomerase-indole-3-glycerol-phosphate synthase from Escherichia coli. Proc Natl Acad Sci USA 1987;84(16):5690-4. doi:10.1073/pnas.84.16.5690
- ^ Hyde C.C., Ahmed S.A., Padlan E.A., Miles E.W., and Davies D.R. Three-dimensional structure of the tryptophan synthase multienzyme complex from Salmonella typhimurium. J Biol Chem 1988:263(33);17857-17871. doi:10.1016/S0021-9258(19)77913-7
- ^ Hyde C.C., Miles E.W. The tryptophan synthase multienzyme complex: exploring structure-function relationships with X-ray crystallography and mutagenesis. Nat Biotechnol 1990:8;27-32. doi:10.1038/nbt0190-27
- ^ Muchmore C.R.A., Krahn J.M, Smith J.L., Kim J.H., Zalkin H. Crystal structure of glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Protein Sci 1998:7;39-51. doi:10.1002/pro.5560070104
- ^ Raushel F.M., Thoden J.B., Holden H.M. The amidotransferase family of enzymes: molecular machines for the production and delivery of ammonia. Biochemistry 1999;38(25):7891-9. doi:10.1021/bi990871p
- ^ Del Caño-Ochoa F., Moreno-Morcillo M., Ramón-Maiques S. CAD, a multienzymatic protein at the head of de novo pyrimidine biosynthesis. Subcell biochem 2019;93:505-538. doi:10.1007/978-3-030-28151-9_17
- ^ Washabaugh M.W., Collins K.D. Dihydroorotase from Escherichia coli. Purification and characterization. J Biol Chem 1984;259(5):3293-8.
- ^ Guan H.H., Huang Y.H., Lin E.S., Chen C.J., Huang C.Y. Structural analysis of Saccharomyces cerevisiae dihydroorotase reveals molecular insights into the tetramerization mechanism. Molecules 2021;26(23):7249. doi:10.3390/molecules26237249
- ^ Belkaïd M., Penverne B., Hervé G. In situ behavior of the pyrimidine pathway enzymes in Saccharomyces cerevisiae. 3. Catalytic and regulatory properties of carbamylphosphate synthetase: channeling of carbamylphosphate to aspartate transcarbamylase. Arch Biochem Biophys 1988;262(1):171-80. doi:10.1016/0003-9861(88)90179-8