

The glucose-alanine cycle, also known as the Cahill cycle, was first described independently by Mallette, Exton, and Park, and by Felig and colleagues between 1969 and 1970. It consists of a series of metabolic steps through which extrahepatic tissues export pyruvate and amino groups to the liver in the form of alanine, while receiving glucose from the liver via the bloodstream.[1][2]
Through this interorgan exchange, the glucose-alanine cycle provides a functional link between carbohydrate and amino acid metabolism, as schematically illustrated below.[3]
Glucose → Pyruvate → Alanine → Pyruvate → Glucose
Although originally described in skeletal muscle, the glucose-alanine cycle is not restricted to muscle–liver interactions, but also involves other extrahepatic tissues, including cells of the immune system.[4]
The glucose-alanine cycle shares functional similarities with the Cori cycle; however, unlike lactate, alanine transports both carbon and nitrogen, thereby directly linking energy metabolism with nitrogen metabolism.[3]
Summary: Key Points
- Definition: a metabolic pathway in which extrahepatic tissues (mainly skeletal muscle) export nitrogen and the carbon skeleton of pyruvate, produced by glycolysis, to the liver in the form of alanine.
- Physiological roles: transport of nitrogen in a non-toxic form and of the gluconeogenic substrate pyruvate to the liver, thereby shifting the metabolic burden associated with nitrogen disposal and glucose regeneration to the liver.
- Energy cost: net energy cost to the liver of approximately 3–5 ATP per cycle, necessary to support gluconeogenesis and the urea cycle.
- Comparison with the Cori cycle: unlike the Cori cycle, it requires oxygen and functional mitochondria and involves the transport of both carbon and nitrogen.
Contents
- Steps of the glucose-alanine cycle
- Mechanisms and biochemical bases
- Physiological roles
- Energy cost of the glucose-alanine cycle
- Similarities with the Cori cycle
- Differences from the Cori cycle
- References
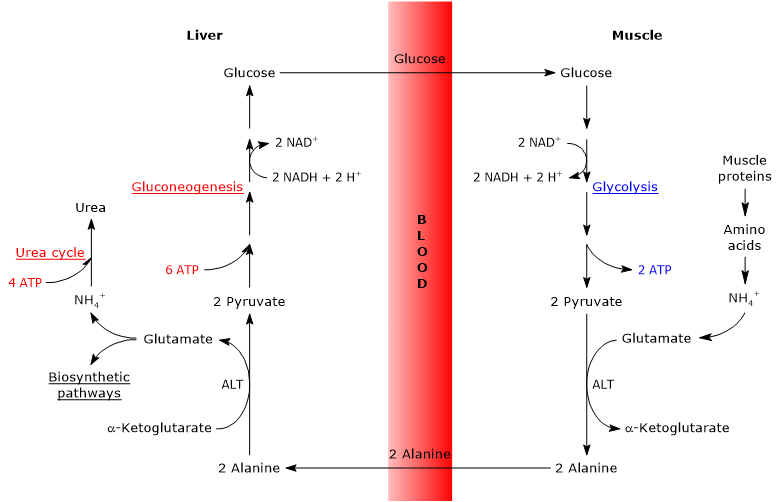
Steps of the glucose-alanine cycle
The principal steps of the glucose-alanine cycle can be outlined as follows.
- When amino acids are oxidized for energy in extrahepatic tissues, pyruvate produced via glycolysis acts as an amino-group acceptor, leading to the formation of alanine, a nonessential amino acid.
- Alanine is released into the bloodstream and transported to the liver.
- In hepatocytes, the amino group of alanine is transferred to α-ketoglutarate, producing pyruvate and glutamate.
- Most of the amino groups derived from glutamate enter the urea cycle, whereas a smaller fraction may serve as nitrogen donors in biosynthetic reactions.
- The pyruvate generated in the liver enters gluconeogenesis and is used for glucose synthesis.
- Newly synthesized glucose is released into the bloodstream and taken up by peripheral tissues, where it is converted back into pyruvate via glycolysis, thereby completing the cycle.

Mechanisms and biochemical bases
The following section examines the biochemical basis of the glucose-alanine cycle, with particular emphasis on the metabolic interplay between skeletal muscle and the liver.
Under physiological conditions, both intracellular and extracellular proteins undergo continuous turnover, being hydrolyzed into their constituent amino acids and subsequently resynthesized. The rates of these opposing processes are tightly regulated, thereby preventing the loss of fat-free mass.[5]
Under catabolic conditions, such as prolonged fasting or intense and sustained exercise, muscle protein breakdown exceeds protein synthesis. This imbalance leads to the release of amino acids, some of which are oxidized for energy production, while others are diverted toward gluconeogenesis.[3]
The oxidation of amino acid carbon skeletons, particularly those of branched-chain amino acids (BCAAs) such as leucine, isoleucine, and valine, can represent a significant energy source for skeletal muscle. Depending on their catabolic fate, amino acids are classified as glucogenic or ketogenic, according to whether their carbon skeletons give rise to substrates for gluconeogenesis or ketone body synthesis, respectively.[6] For example, after approximately 90 minutes of strenuous exercise, amino acid oxidation may account for 10–15% of the total energy required for muscle contraction.[7]
The use of amino acid carbon skeletons for energy necessarily involves the removal of their amino groups, followed by the safe excretion of amino nitrogen in a non-toxic form.[8]
Removal of the α-amino group occurs primarily through transamination, which can be summarized by the following reversible reaction:
α-Keto acid + Amino acid ⇌ New amino acid + New α-keto acid
These reactions, catalyzed by enzymes known as aminotransferases or transaminases (EC 2.6.1.-), are freely reversible.[9]
In skeletal muscle, branched-chain amino acids commonly transfer their amino group to α-ketoglutarate (2-oxoglutarate), forming glutamate and the corresponding branched-chain α-keto acids. This reaction is catalyzed by branched-chain aminotransferase (BCAT, EC 2.6.1.42).[10]
Glucose-alanine cycle in skeletal muscle
In skeletal muscle, newly formed glutamate may react with ammonia to form glutamine, which serves as a major vehicle for the interorgan transport of nitrogen, particularly to tissues such as the brain.[11] This reaction is catalyzed by the cytosolic enzyme glutamine synthetase (EC 6.3.1.2) and requires the consumption of one molecule of ATP:
Glutamate + NH4+ + ATP → Glutamine + ADP + Pi
In this case, glutamate exits the Cahill cycle.[9]
Alternatively, and in contrast to what occurs in most other tissues, glutamate may transfer its amino group to pyruvate (derived from glycolysis) to form alanine and α-ketoglutarate. This transamination is catalyzed by alanine aminotransferase (ALT, EC 2.6.1.2), an enzyme widely distributed in both animal and plant tissues:[12]
Glutamate + Pyruvate ⇄ Alanine + α-Ketoglutarate
The alanine thus produced, along with that released directly from muscle protein breakdown (muscle proteins are relatively rich in alanine), can exit the cell and enter the bloodstream, ultimately reaching the liver. In this way, the amino group is efficiently transported to the liver.[13]
Moreover, the rate at which alanine formed by pyruvate transamination enters the circulation is proportional to intracellular pyruvate production.[14]
Note: alanine and glutamine are the major interorgan carriers of nitrogen and carbon derived from amino acid metabolism.[3]
Glucose-alanine cycle in the liver
Once in the liver, hepatic alanine aminotransferase catalyzes a transamination in which alanine, the major gluconeogenic amino acid, acts as an amino group donor, while α-ketoglutarate serves as the α-keto acid acceptor.[15] The products of this reaction are pyruvate (the carbon skeleton of alanine) and glutamate:
Alanine + α-Ketoglutarate ⇄ Glutamate + Pyruvate
In a subsequent reaction catalyzed by glutamate dehydrogenase (EC 1.4.1.2), an enzyme located in the mitochondrial matrix, glutamate is deaminated to produce an ammonium ion (NH4+), which enters the urea cycle, and α-ketoglutarate, which may re-enter the citric acid cycle. This reaction is considered anaplerotic, as it links amino acid metabolism with the citric acid cycle.[16]
Glutamate + H2O + NAD+ ⇄ α-Ketoglutarate + NH4+ + NADH + H+
Alternatively, glutamate can undergo transamination with oxaloacetate to form aspartate and α-ketoglutarate, in a reaction catalyzed by aspartate aminotransferase (EC 2.6.1.1).[17] Aspartate is not only involved in urea synthesis, but also plays a key role in the biosynthesis of purines and pyrimidines.[18]
Glutamate + Oxaloacetate ⇄ Aspartate + α-Ketoglutarate
The pyruvate generated from alanine may follow two alternative metabolic fates.
- It can be oxidized for ATP production, in which case it exits the glucose-alanine cycle.
- It can enter the gluconeogenesis pathway, continuing the cycle.[19]
The glucose synthesized in the liver is released into the bloodstream and delivered to peripheral tissues, such as skeletal muscle, where it is converted into pyruvate through glycolysis. This newly formed pyruvate may then accept an amino group from glutamate, thus completing the cycle.[20]
Transaminases
As noted above, the removal of amino groups from amino acids occurs via transamination. These reactions are catalyzed by enzymes known as aminotransferases or transaminases.[12]
Transaminases are cytosolic enzymes present in all cells, but are particularly abundant in the liver, kidney, intestine, and muscle. They require pyridoxal phosphate (PLP), the active form of vitamin B6 (pyridoxine), as a tightly bound coenzyme at their active site.[13]
During transamination, the amino group of most free amino acids (with the exceptions of threonine and lysine) is transferred to one of a small set of keto acids, primarily pyruvate, oxaloacetate, or α-ketoglutarate.[3]
Cells express multiple aminotransferases that share specificity for α-ketoglutarate as the amino-group acceptor but differ in their amino-acid substrates, from which they derive their names. Key examples include:
- alanine aminotransferase, also known as alanine transaminase or glutamate-pyruvate transaminase (GPT);
- aspartate aminotransferase (AST), also called glutamate-oxaloacetate transaminase (GOT).[9]
Importantly, transamination reactions do not result in net deamination; the amino group simply “swaps” from the amino acid to the keto acid, with no loss of amino groups overall.[12]
Physiological roles
The glucose-alanine cycle serves several important physiological functions.
- It transports nitrogen in a non-toxic form from peripheral tissues to the liver in the form of alanine.[8]
- It transports pyruvate, a key gluconeogenic substrate, to the liver for glucose synthesis.[8]
- It removes pyruvate from peripheral tissues, promoting more efficient ATP production. By reducing cytosolic pyruvate, the NADH generated during glycolysis can be more effectively oxidized via mitochondrial oxidative phosphorylation.[13]
- It helps maintain a relatively high concentration of alanine in hepatocytes, which may contribute to the inhibition of hepatic protein degradation.[15]
- It may contribute to host defense mechanisms during infectious diseases, although the precise role is still under investigation.[21]
| Function | Description |
|---|---|
| Nitrogen transport | Safely carries nitrogen from peripheral tissues to the liver in the form of alanine. |
| Pyruvate transport | Delivers pyruvate (a gluconeogenic substrate) to the liver for glucose production. |
| ATP optimization | Enhances ATP yield in peripheral tissues by enabling full oxidation of glycolytic NADH via mitochondria. |
| Protein-sparing effect | Helps maintain high alanine levels in hepatocytes, potentially reducing hepatic protein degradation. |
| Support in host defense | May contribute to immune response during infections (mechanism under study). |
| No net glucose gain | The cycle does not result in net glucose synthesis. |
Finally, it is important to emphasize that there is no net synthesis of glucose in the glucose-alanine cycle.[3]
Energy cost of the glucose-alanine cycle
Like the Cori cycle, the glucose-alanine cycle incurs an energy cost, estimated at approximately 3–5 ATP per cycle.[22]
The peripheral (extrahepatic) part of the cycle yields energy:
- 2 ATP are generated through glycolysis.
- An additional 3–5 ATP are produced from the oxidation of NADH and FADH2.
In contrast, the hepatic part of the cycle (in the liver) consumes energy:
- 6 ATP are required for gluconeogenesis per molecule of glucose synthesized;
- 4 ATP are used in the urea cycle per molecule of urea formed.
Thus, the glucose-alanine cycle, like the Cori cycle, shifts part of the metabolic burden from extrahepatic tissues to the liver. However, the energy cost sustained by the liver is justified by the physiological benefits the cycle provides. It facilitates the breakdown of proteins in extrahepatic tissues, particularly skeletal muscle, under specific conditions (e.g., prolonged exercise or fasting). This, in turn, enables both the utilization of amino acids for energy and the supply of gluconeogenic substrates to the liver.[3]
Similarities with the Cori cycle
There are several similarities between the two cycles, summarized below.
- The Cahill cycle partially overlaps with the Cori cycle when pyruvate is converted into glucose in the liver, and the resulting glucose is transported back to extrahepatic tissues, where it is reconverted into pyruvate via glycolysis.[3]
- The entry point into the gluconeogenic pathway is similar in both cycles: both alanine and lactate are converted to pyruvate.[9]
- Like the Cori cycle, the glucose-alanine cycle occurs between different cell types, unlike metabolic pathways such as glycolysis, the citric acid cycle, or gluconeogenesis, which take place within individual cells.[23]

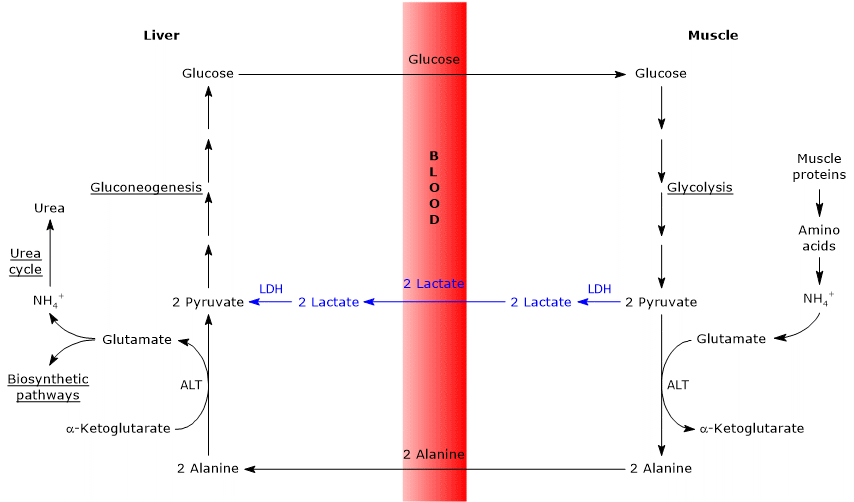
Differences from the Cori cycle
Here are some key differences between the two cycles.
- The main difference lies in the three-carbon intermediate transported from peripheral tissues to the liver: lactate in the Cori cycle, and alanine in the glucose-alanine cycle.[15]
- Another difference involves the fate of NADH produced by glycolysis in peripheral tissues.
- In the Cori cycle, NADH is used to reduce pyruvate to lactate via the enzyme lactate dehydrogenase (EC 1.1.1.27).
- In the glucose-alanine cycle, this reduction does not occur. Instead, electrons from NADH are transferred into the mitochondria via the malate–aspartate and glycerol-3-phosphate shuttles, generating NADH through the former and FADH2 through the latter. The ATP yields are approximately 2.5 per NADH and 1.5 per FADH2.[23]
- As a result, unlike the Cori cycle, the glucose-alanine (Cahill) cycle requires the presence of both oxygen and functional mitochondria in peripheral tissues.[11]
| Feature | Cori cycle | Glucose-alanine cycle |
|---|---|---|
| Three-carbon compound transported to liver | Lactate | Alanine |
| Source of 3-carbon compound | Reduction of pyruvate by NADH | Transamination of pyruvate with glutamate |
| NADH usage in peripheral tissues | Consumed to reduce pyruvate to lactate | Transferred to mitochondria via shuttles |
| Requirement for oxygen and mitochondria | Not required | Required |
| Nitrogen transport | None | Transports amino groups (as alanine) to liver |
| Occurs between different cell types | Yes | Yes |
| Entry into gluconeogenesis | Lactate → Pyruvate | Alanine → Pyruvate |
| Energy cost in liver | High (gluconeogenesis) | High (gluconeogenesis + urea cycle) |
References
- ^ Felig P., Pozefsky T., Marlis E., Cahill G.F. Alanine: key role in gluconeogenesis. Science 1970;167(3920):1003-1004. doi:10.1126/science.167.3920.1003
- ^ Mallette L.E., Exton J.H., and Park C.R. Control of gluconeogenesis from amino acids in the perfused rat liver. J Biol Chem 1969;244(20):5713-5723. doi:10.1016/S0021-9258(18)63618-X
- ^ a b c d e f g h Nelson D.L., Cox M.M. Lehninger. Principles of biochemistry. 8th Edition. W.H. Freeman and Company, 2021.
- ^ Zhao H., Raines L.N., Huang S.C. Carbohydrate and amino acid metabolism as hallmarks for innate immune cell activation and function. Cells 2020;9(3):562. doi:10.3390/cells9030562
- ^ Lecker S.H., Goldberg A.L. and Mitch W.E. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J Am Soc Nephrol 2006;17(7):1807-1819. doi:10.1681/ASN.2006010083
- ^ Rennie M.J., Tipton K.D. Protein and amino acid metabolism during and after exercise and the effects of nutrition. Annu Rev Nutr 2000;20:457-83. doi:10.1146/annurev.nutr.20.1.457
- ^ Witard O.C., Hearris M., Morgan P.T. Protein nutrition for endurance athletes: a metabolic focus on promoting recovery and training adaptation. Sports Med 2025;55(6):1361-1376. doi:10.1007/s40279-025-02203-8
- ^ a b c Moran L.A., Horton H.R., Scrimgeour K.G., Perry M.D. Principles of Biochemistry. 5th Edition. Pearson, 2012.
- ^ a b c d Heilman D., Woski S., Voet D., Voet J.G., Pratt C.W. Fundamentals of biochemistry: life at the molecular level. 6th Edition. Wiley, 2023.
- ^ Nong X., Zhang C., Wang J., Ding P., Ji G., Wu T. The mechanism of branched-chain amino acid transferases in different diseases: research progress and future prospects. Front Oncol 2022;12:988290. doi:10.3389/fonc.2022.988290
- ^ a b Wu G. Amino acids: biochemistry and nutrition. CRC Press, 2010.
- ^ a b c Berg J.M., Tymoczko J.L., Gatto J.G., Stryer L. Biochemistry. 9th Edition. W.H. Freeman and Company, 2019.
- ^ a b c Rodwell V.W., Bender D.A., Botham K.M., Kennelly P.J., Weil P.A. Harper’s illustrated biochemistry. 31st Edition. McGraw-Hill, 2018.
- ^ Owen O.E., Morgan A.P., Kemp H.G., Sullivan J.M., Herrera M.G., Cahill G.F. Jr. Brain metabolism during fasting. J Clin Invest 1967;46(10):1589-95. doi:10.1172/JCI105650
- ^ a b c Felig P. The glucose-alanine cycle. Metabolism 1973;22(2):179-207. doi:10.1016/0026-0495(73)90269-2
- ^ Rosenthal M.D., Glew R.H. Medical biochemistry – Human metabolism in health and disease. John Wiley & Sons, 2009.
- ^ Chandel N.S. Amino acid metabolism. Cold Spring Harb Perspect Biol 2021;13(4):a040584. doi:10.1101/cshperspect.a040584
- ^ Kurmi K., Haigis M.C. Nitrogen metabolism in cancer and immunity. Trends Cell Biol 2020;30(5):408-424. doi:10.1016/j.tcb.2020.02.005
- ^ Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci 2014;71(14):2577-604. doi:10.1007/s00018-013-1539-2
- ^ Petersen K.F., Dufour S., Cline G.W., Shulman G.I. Regulation of hepatic mitochondrial oxidation by glucose-alanine cycling during starvation in humans. J Clin Invest 2019;129(11):4671-4675. doi:10.1172/JCI129913
- ^ Newsholme E.A., Parry-Billings M. Properties of glutamine release from muscle and its importance for the immune system. JPEN J Parenter Enteral Nutr 1990;14(4 Suppl):63S-67S. doi:10.1177/014860719001400406
- ^ Felig P. Amino acid metabolism in man. Annu Rev Biochem 1975;44:933-55. doi:10.1146/annurev.bi.44.070175.004441
- ^ a b Garrett R.H., Grisham C.M. Biochemistry. 7th Edition. Cengage Learning, 2023.